当前位置:网站首页>Scanpy (VI) analysis and visualization of spatial transcriptome data
Scanpy (VI) analysis and visualization of spatial transcriptome data
2022-06-12 15:56:00 【tzc_ fly】
Catalog
This article demonstrates how to Scanpy Using spatial transcriptome data in (spatial transcriptomics data), First , We focus on 10x Genomics Visium data, Last , We are MERFISH Technology provides an example of spatial transcription data analysis .
At present, the technology of spatial transcriptome has not reached the level of single cell , A general spatial location spots There are multiple cells on the ,10X Genomics Visum It's usually 1-10 Cells , Spatial transcriptomics can provide spatial location , But the resolution can not reach the single cell level , Single cell transcriptome resolution can reach single cell resolution , But there is no spatial location information , Therefore, if the spatial transcriptome is combined with the single-cell transcriptome , Then there is the spatial location information , The single cell resolution is reached , This provides a powerful tool for the scientific community , Greatly improve the organization of organisms 、 The resolution and accuracy of organ studies .
We import scanpy Tools :
import scanpy as sc
import pandas as pd
import matplotlib.pyplot as plt
import seaborn as sns
sc.logging.print_versions()
sc.set_figure_params(facecolor="white", figsize=(8, 8))
sc.settings.verbosity = 3
""" anndata 0.8.0 scanpy 1.9.1 """
Reading data
We will use human lymph nodes (human lymphnode) Of Visium Spatial transcriptomics data set , This data set is published in 10x genomics website:link;
function datasets.visium_sge() from 10x Genomics Download datasets , And returns a matrix containing the count counts And spatial coordinates spatital coordinates Of AnnData object .
adata = sc.datasets.visium_sge(sample_id="V1_Human_Lymph_Node")
adata.var_names_make_unique()
adata
""" AnnData object with n_obs × n_vars = 4035 × 36601 obs: 'in_tissue', 'array_row', 'array_col' var: 'gene_ids', 'feature_types', 'genome' uns: 'spatial' obsm: 'spatial' """
We will use pp.calculate_qc_metrics And based on mitochondria read counts Calculation QC indicators .
# Save the mitochondrial genome as an annotation var.mt
adata.var["mt"] = adata.var_names.str.startswith("MT-")
adata.var["mt"]
""" MIR1302-2HG False ... AC007325.2 False Name: mt, Length: 36601, dtype: bool """
# Calculate the index , qc Of var choice var.mt
sc.pp.calculate_qc_metrics(adata, qc_vars=["mt"], inplace=True)
adata
""" AnnData object with n_obs × n_vars = 4035 × 36601 obs: 'in_tissue', 'array_row', 'array_col', 'n_genes_by_counts', 'log1p_n_genes_by_counts', 'total_counts', 'log1p_total_counts', 'pct_counts_in_top_50_genes', 'pct_counts_in_top_100_genes', 'pct_counts_in_top_200_genes', 'pct_counts_in_top_500_genes', 'total_counts_mt', 'log1p_total_counts_mt', 'pct_counts_mt' var: 'gene_ids', 'feature_types', 'genome', 'mt', 'n_cells_by_counts', 'mean_counts', 'log1p_mean_counts', 'pct_dropout_by_counts', 'total_counts', 'log1p_total_counts' uns: 'spatial' obsm: 'spatial' """
review Scanpy( 3、 ... and ) Handle 3k PBMCs Parallel clustering The content in , We can understand the meaning of some quality :
n_genes_by_counts: In every cell , Number of genes expressed ;total_counts: Total gene count per cell ( Total meter volume );pct_counts_mt: In every cell , The percentage of mitochondrial gene expression in all gene expression of the cell ;
QC and preprocessing
We according to the total_counts and n_genes_by_counts Do some basic filtering . We first visualize these quality indicators as histograms :
fig, axs = plt.subplots(1, 4, figsize=(15, 4))
sns.distplot(adata.obs["total_counts"], kde=False, ax=axs[0])
sns.distplot(adata.obs["total_counts"][adata.obs["total_counts"] < 10000], kde=False, bins=40, ax=axs[1])
sns.distplot(adata.obs["n_genes_by_counts"], kde=False, bins=60, ax=axs[2])
sns.distplot(adata.obs["n_genes_by_counts"][adata.obs["n_genes_by_counts"] < 4000], kde=False, bins=60, ax=axs[3])
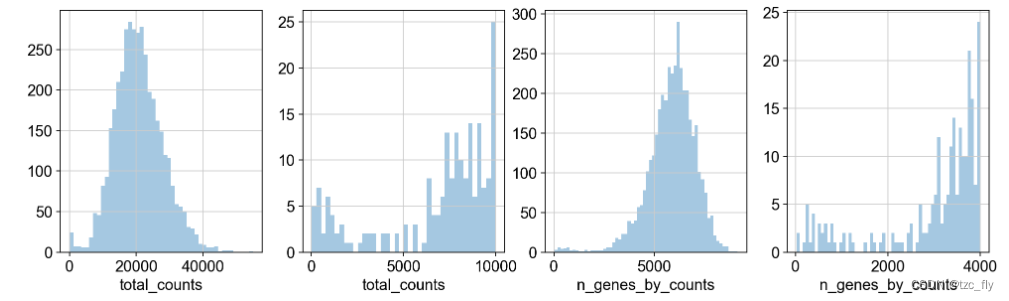
The second graph is the first graph in 0 To 10000 Visualization of intervals ,total_counts Represents the total gene expression of a cell . The fourth graph is the third graph in 0 To 4000 Visualization ,n_genes_by_counts Represents a cell , Number of genes expressed . Be careful , The counting matrix is 4035×36601.
Through the above visualization results , We reserve the total_counts stay 5000 To 35000 The cells , next step , We keep mitochondrial genes pct_counts_mt The proportion is less than 20% The cells . Last , Do genetic filtering :
sc.pp.filter_cells(adata, min_counts=5000)
sc.pp.filter_cells(adata, max_counts=35000)
adata = adata[adata.obs["pct_counts_mt"] < 20]
print(f"#cells after MT filter: {
adata.n_obs}")
sc.pp.filter_genes(adata, min_cells=10)
""" filtered out 44 cells that have less than 5000 counts filtered out 130 cells that have more than 35000 counts #cells after MT filter: 3861 filtered out 16916 genes that are detected in less than 10 cells """
adata
""" AnnData object with n_obs × n_vars = 3861 × 19685 obs: 'in_tissue', 'array_row', 'array_col', 'n_genes_by_counts', 'log1p_n_genes_by_counts', 'total_counts', 'log1p_total_counts', 'pct_counts_in_top_50_genes', 'pct_counts_in_top_100_genes', 'pct_counts_in_top_200_genes', 'pct_counts_in_top_500_genes', 'total_counts_mt', 'log1p_total_counts_mt', 'pct_counts_mt', 'n_counts' var: 'gene_ids', 'feature_types', 'genome', 'mt', 'n_cells_by_counts', 'mean_counts', 'log1p_mean_counts', 'pct_dropout_by_counts', 'total_counts', 'log1p_total_counts', 'n_cells' uns: 'spatial' obsm: 'spatial' """
We continue to use scanpy Built in functions for normaliza_total Standardization Visium counts data( contain counts And spatially transcribed information ), And then detect hypervariable genes .
sc.pp.normalize_total(adata, inplace=True)
sc.pp.log1p(adata)
sc.pp.highly_variable_genes(adata, flavor="seurat", n_top_genes=2000)
""" normalizing counts per cell finished (0:00:00) If you pass `n_top_genes`, all cutoffs are ignored. extracting highly variable genes finished (0:00:00) --> added 'highly_variable', boolean vector (adata.var) 'means', float vector (adata.var) 'dispersions', float vector (adata.var) 'dispersions_norm', float vector (adata.var) """
adata
""" AnnData object with n_obs × n_vars = 3861 × 19685 obs: 'in_tissue', 'array_row', 'array_col', 'n_genes_by_counts', 'log1p_n_genes_by_counts', 'total_counts', 'log1p_total_counts', 'pct_counts_in_top_50_genes', 'pct_counts_in_top_100_genes', 'pct_counts_in_top_200_genes', 'pct_counts_in_top_500_genes', 'total_counts_mt', 'log1p_total_counts_mt', 'pct_counts_mt', 'n_counts' var: 'gene_ids', 'feature_types', 'genome', 'mt', 'n_cells_by_counts', 'mean_counts', 'log1p_mean_counts', 'pct_dropout_by_counts', 'total_counts', 'log1p_total_counts', 'n_cells', 'highly_variable', 'means', 'dispersions', 'dispersions_norm' uns: 'spatial', 'log1p', 'hvg' obsm: 'spatial' """
Manifold embedding and clustering based on transcriptional similarity
First , We follow the standard clustering procedure (pca Dimension reduction , Calculation neighbors graph,umap Dimension reduction ,leiden clustering ):
sc.pp.pca(adata)
sc.pp.neighbors(adata)
sc.tl.umap(adata)
sc.tl.leiden(adata, key_added="clusters")
""" computing PCA on highly variable genes with n_comps=50 finished (0:00:01) computing neighbors using 'X_pca' with n_pcs = 50 finished: added to `.uns['neighbors']` `.obsp['distances']`, distances for each pair of neighbors `.obsp['connectivities']`, weighted adjacency matrix (0:00:06) computing UMAP finished: added 'X_umap', UMAP coordinates (adata.obsm) (0:00:06) running Leiden clustering finished: found 10 clusters and added 'clusters', the cluster labels (adata.obs, categorical) (0:00:00) """
adata
""" AnnData object with n_obs × n_vars = 3861 × 19685 obs: 'in_tissue', 'array_row', 'array_col', 'n_genes_by_counts', 'log1p_n_genes_by_counts', 'total_counts', 'log1p_total_counts', 'pct_counts_in_top_50_genes', 'pct_counts_in_top_100_genes', 'pct_counts_in_top_200_genes', 'pct_counts_in_top_500_genes', 'total_counts_mt', 'log1p_total_counts_mt', 'pct_counts_mt', 'n_counts', 'clusters' var: 'gene_ids', 'feature_types', 'genome', 'mt', 'n_cells_by_counts', 'mean_counts', 'log1p_mean_counts', 'pct_dropout_by_counts', 'total_counts', 'log1p_total_counts', 'n_cells', 'highly_variable', 'means', 'dispersions', 'dispersions_norm' uns: 'spatial', 'log1p', 'hvg', 'pca', 'neighbors', 'umap', 'leiden' obsm: 'spatial', 'X_pca', 'X_umap' varm: 'PCs' obsp: 'distances', 'connectivities' """
We are based on umap Visualize some related variables , To check for presence and total count (total_counts) And detected genes (n_genes_by_counts) Relevant specific data distribution :
plt.rcParams["figure.figsize"] = (4, 4)
sc.pl.umap(adata, color=["total_counts", "n_genes_by_counts", "clusters"], wspace=0.4)

Visualization in spatial coordinates
We can look at the space coordinates first :
adata.obsm['spatial'].shape
# (3861, 2)
adata.obsm['spatial']
""" array([[8346, 6982], [4270, 1363], ..., [4822, 1840]], dtype=int64) """
Now let's see total_counts and n_genes_by_counts Distribution in spatial coordinates . We will use sc.pl.spatial The function returns a circular dot (circular spots) Cover the provided Hematoxylin and eosin stain(H&E) On the image .
plt.rcParams["figure.figsize"] = (8, 8)
sc.pl.spatial(adata, img_key="hires", color=["total_counts", "n_genes_by_counts"])

function sc.pl.spatial Accept 4 Additional parameters :
img_key: Stored inadata.unsinimagesInsidekey;crop_coord: Coordinates for cropping (left, right, top, bottom);alpha_img: transparency ;bw: A sign that converts an image to a grayscale image ;
Besides , stay sc.pl.spatial in ,size Parameters change their behavior : It will become spots Scale factor of size .
Before , We cluster in the gene expression space , And use UMAP Visualize results . Now let's visualize the sample in the spatial dimension , We can gain insight into the structure of the organization , And may have a deep understanding of the communication between cells :
sc.pl.spatial(adata, img_key="hires", color="clusters", size=1.5)

Belonging to the same cluster in the gene expression space spots It usually appears in the spatial dimension at the same time . for example , Belong to cluster 4 Points of are usually classified as clusters 0 Surrounded by points .
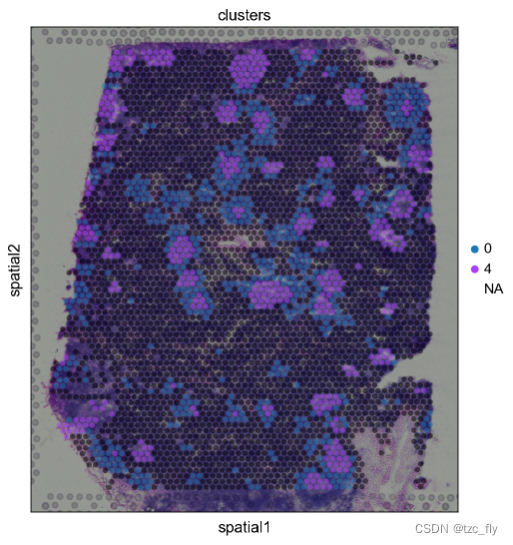
We can zoom in on a particular region of interest , To reach a particular conclusion . Besides , By changing spot Of alpha value , We can better learn from H&E Potential tissue morphology seen in the image .
sc.pl.spatial(adata, img_key="hires", color="clusters", groups=["0", "4"], alpha=0.5, size=1.3)
Clustered marker gene
Let's examine the cluster further 4. We calculate the marker genes and draw a heat map , The figure shows the front... In the cluster 10 Expression levels of marker genes .
sc.tl.rank_genes_groups(adata, "clusters", method="t-test")
sc.pl.rank_genes_groups_heatmap(adata, groups="4", n_genes=10, groupby="clusters")
""" ranking genes finished: added to `.uns['rank_genes_groups']` 'names', sorted np.recarray to be indexed by group ids 'scores', sorted np.recarray to be indexed by group ids 'logfoldchanges', sorted np.recarray to be indexed by group ids 'pvals', sorted np.recarray to be indexed by group ids 'pvals_adj', sorted np.recarray to be indexed by group ids (0:00:02) WARNING: dendrogram data not found (using key=dendrogram_clusters). Running `sc.tl.dendrogram` with default parameters. For fine tuning it is recommended to run `sc.tl.dendrogram` independently. using 'X_pca' with n_pcs = 50 Storing dendrogram info using `.uns['dendrogram_clusters']` WARNING: Groups are not reordered because the `groupby` categories and the `var_group_labels` are different. categories: 0, 1, 2, etc. var_group_labels: 4 """

We are interested in genes CR2 Analyze :
sc.pl.spatial(adata, img_key="hires", color=["clusters", "CR2"])

Hypervariable genes of spatial data
Spatial transcriptomics enables researchers to study changes in gene expression trends in space , So as to determine the spatial pattern of gene expression . So , We use SpatialDE, This is a statistical framework based on Gaussian process , Variable genes designed to identify spatial transcriptome :
pip install spatialde
lately , Other tools for identifying spatially variant genes have also been proposed , for example :SPARK,trendsceek,HMRF
First , We will standardize counts and coordinates Convert to pandas dataframe, This is the input spatialDE The required :
import SpatialDE
%%time
counts = pd.DataFrame(adata.X.todense(), columns=adata.var_names, index=adata.obs_names)
coord = pd.DataFrame(adata.obsm['spatial'], columns=['x_coord', 'y_coord'], index=adata.obs_names)
""" CPU times: total: 93.8 ms Wall time: 94.7 ms """
see counts:
see coord:
function SpatialDE It takes quite a long time ( This example is close to 2 Hours ).
results = SpatialDE.run(coord, counts)
results The variable genes calculated based on the spatial transcriptome data will be saved in .
MERFISH Example
If we use based on FISH Technology to generate spatial data , Just read cordinate Table and assign it to adata.obsm that will do .
Let's look at Xia wait forsomeone 2019 The example of .
First , We need to download coordinates and counts data :
import urllib.request
url_coord = "https://www.pnas.org/highwire/filestream/887973/field_highwire_adjunct_files/15/pnas.1912459116.sd15.xlsx"
filename_coord = "pnas.1912459116.sd15.xlsx"
urllib.request.urlretrieve(url_coord, filename_coord)
url_counts = "https://www.pnas.org/highwire/filestream/887973/field_highwire_adjunct_files/12/pnas.1912459116.sd12.csv"
filename_counts = "pnas.1912459116.sd12.csv"
urllib.request.urlretrieve(url_counts, filename_counts)
If you can't access the above link , You can manually go to github download :spatial datasets, This is the collation of a spatial transcription data set .
And then it reads adata:
coordinates = pd.read_excel("./pnas.1912459116.sd15.xlsx", index_col=0)
counts = sc.read_csv("./pnas.1912459116.sd12.csv").transpose()
adata_merfish = counts[coordinates.index, :]
adata_merfish.obsm["spatial"] = coordinates.to_numpy()
adata_merfish
""" AnnData object with n_obs × n_vars = 645 × 12903 obsm: 'spatial' """
We will perform standard preprocessing and dimensionality reduction :
sc.pp.normalize_per_cell(adata_merfish, counts_per_cell_after=1e6)
sc.pp.log1p(adata_merfish)
sc.pp.pca(adata_merfish, n_comps=15)
sc.pp.neighbors(adata_merfish)
sc.tl.umap(adata_merfish)
sc.tl.leiden(adata_merfish, key_added="clusters", resolution=0.5)
""" normalizing by total count per cell finished (0:00:00): normalized adata.X and added 'n_counts', counts per cell before normalization (adata.obs) computing PCA with n_comps=15 finished (0:00:00) computing neighbors using 'X_pca' with n_pcs = 15 finished: added to `.uns['neighbors']` `.obsp['distances']`, distances for each pair of neighbors `.obsp['connectivities']`, weighted adjacency matrix (0:00:05) computing UMAP finished: added 'X_umap', UMAP coordinates (adata.obsm) (0:00:01) running Leiden clustering finished: found 7 clusters and added 'clusters', the cluster labels (adata.obs, categorical) (0:00:00) """
adata_merfish
""" AnnData object with n_obs × n_vars = 645 × 12903 obs: 'n_counts', 'clusters' uns: 'log1p', 'pca', 'neighbors', 'umap', 'leiden' obsm: 'spatial', 'X_pca', 'X_umap' varm: 'PCs' obsp: 'distances', 'connectivities' """
We can do it in UMAP Space and spatial Visualize in coordinates Leiden Resulting cluster .
sc.pl.umap(adata_merfish, color="clusters")
sc.pl.embedding(adata_merfish, basis="spatial", color="clusters")

边栏推荐
- redis String类型常见命令
- Saga architecture pattern: implementation of cross service transactions under microservice architecture
- 一步步创建包含自定义 Screen 的 ABAP 程序的详细步骤
- 为什么阿里巴巴不建议MySQL使用Text类型?
- Remote control of other computers -- detailed tutorial
- Job submission instructions upload jobs to network disk
- glibc 内存管理模型 释放 C库内存缓存
- 同源?跨域?如何解决跨域?
- The nohup command uses
- TCP与UDP的区别,以及TCP的三次握手和TCP的四次挥手
猜你喜欢

广播和多播(TCP/IP详解卷1/2)

Golang collaboration scheduling (I): Collaboration Status

Dart typedef的理解

< 山东大学软件学院项目实训 > 渲染引擎系统——基础渲染器(三)

Introduction and download website of common data of GIS, remote sensing, hydrology and Geography (2), supplementary~

Classic case of solidity - Smart games

Saga architecture pattern: implementation of cross service transactions under microservice architecture

POSTMAN-REST Client插件的应用

Project training of Software College of Shandong University rendering engine system radiation pre calculation (VIII)

Getting started with JMeter
随机推荐
< 山东大学软件学院项目实训 > 渲染引擎系统——辐射预计算(九)
Import and export steps of SQL Server 2008
< 山东大学软件学院项目实训 > 渲染引擎系统——基础渲染器(三)
Example of bit operation (to be continued)
UE4 常用类型转换
Application of postman-rest client plug-in
任务 水果炸汁机 0611
Understanding of dart typedef
Job submission instructions upload jobs to network disk
FPGA (III) trigger and latch
Two ways of array simulating queue
Codeforces Round 797 (Div. 3,CF1690)全题解
作業提交說明 上傳作業到網盤中
File uploading and downloading in SSM
Fiddler packet capturing (mobile app)
[jvm learning] local method stack and heap
The common hand, the original hand and the excellent hand from the sum of Fibonacci sequence
IDEA中文棱形乱码错误解决方法--控制台中文输出棱形乱码
jupyter notebook新環境快捷方式
Apache Kylin 历险记