当前位置:网站首页>Paper reading (56):muti features predction of protein translational modification sites (task)
Paper reading (56):muti features predction of protein translational modification sites (task)
2022-06-23 18:04:00 【Inge】
List of articles
1 introduce
1.1 subject
1.2 summary
Post translational modification (Post translational modification, PTM) It plays an important role in biological processing . Potential post-translational modifications consist of central sites and adjacent amino acid residues , They are basic protein sequence residues , It helps to exert their biological functions , It is also helpful to understand the molecular mechanism that is the basis of protein design and drug design . The existing modification site prediction algorithms often have low stability and accuracy And so on .
This paper combines the physics of protein 、 chemical 、 Statistical and biological characteristics , A new framework is proposed to predict the post-translational modification sites of proteins . call Multilayer neural network and support vector machine To predict potential modification sites with selected characteristics , These characteristics include the composition of amino acid residues 、 Of protein fragments E-H Description and AAIndex Several properties in the database . Consider possible redundant information , In the processing step, the feature selection . Experimental results show that , The proposed method can improve the accuracy of the classification problem .
1.3 Bib
@article{
Bao:2017:14531460,
author = {
Wen Zheng Bao and Chang-An Yuan and You Hua Zhang and Kyungsook Han and Asoke K Nandi and Barry Honig and De-Shuang Huang},
title = {
Mutli-features prediction of protein translational modification sites},
journal = {
{
IEEE}/{
ACM} Transactions on Computational Biology and Bioinformatics},
volume = {
15},
number = {
5},
pages = {
1453--1460},
year = {
2017},
doi = {
10.1109/TCBB.2017.2752703}
}
2 Method
2.1 Data sets
The function of a protein depends on its spatial conformation . therefore , The spatial structure of protein fragments may be helpful to analyze and identify the characteristics of potential modification sites .
Experimental data sets yes PTM Benchmark data set for the prediction field :
1) A well-known database in the field of protein post-translational modification CPLM. The database contains 2500 Multiple lysine succinylation sites and as positive samples 24000 Non succinylation sites as negative samples , From 896 Protein sequences . All the above protein fragments and polypeptide sequences are from UniProt, This is a famous protein database in the field of bioinformatics . It has been used for enzyme specificity (ES) And protein - Protein binding sites (PPB) The study of .
2) be used for Predict a variety of protein sequences K-PTM Type of modification site Framework , It contains 6394 Potential modification sites , These loci are considered to come from 27 Tuple peptide like . Yes 1750 Samples do not belong to the four K-PTM Any one of the types ,3895 Samples belong to a kind of K-PTM,740 The samples belong to two kinds PTM type ,9 The samples belong to three kinds PTM type , All four types do not .
3) Post - translational modification of fragment data sets . Lysine acetylation site datasets for three species , Including Homo sapiens 、 House mouse and Saccharomyces cerevisiae , From multiple sources , Include PhosphoSite、UniProtKB/Swiss-Prot、UbiProt and SCUD, These are well-known databases in the field of proteomics . Because ubiquitin seems to be attached to lysine residues of proteins to some extent . therefore , In our work, we only considered lysine ubiquitination in the above three species . The original data set includes 11547 Protein sequences covering different species ; In these sequences , exceed 8000 One from H.sapiens, about 3300 One from M.musculus, exceed 4500 One from S.cerevisiae. Remove 3 After the redundant protein fragments of the samples , Extract to 3 Multiple samples of samples , Among them are 6323 Share H.sapiens sample 、2342 Share M.musculus Samples and 7863 Share S.cerevisiaes sample . after , Randomly selected from each data set of three species 20 Three proteins form a separate test set , The rest 6303、2322 and 7843 Three proteins were used to construct the training set .
2.2 feature selection
Generally speaking , The types of protein characteristics can reach 4 More than ten thousand . These various types of features , Including amino acid composition model (AAC) Pseudo amino acid composition model (PseAAC) And other relevant information about protein characteristics [26]. However , These characteristics are difficult to effectively and accurately describe the interaction between predicted modification sites and adjacent amino acid residues . therefore , This paper introduces a typical 、 Special features , It has the ability to describe protein peptides .
First , When it comes to the composition of amino acid residues , Many researchers in bioinformatics and computational biology usually use the statistical information of protein sequences . These characteristics only describe the potential modification of the statistical aspects . Of course , In such feature sets , The selection of key features can be seen as a difficult task .
Found to have 20 Amino acid residues in 3 Class special structural elements : screw 、 There is a tendency to be swallowed up in chains and spirals . These functions are selected from PSIPRED. PSIPRED The developers of e.g .
Consider effectively α \alpha α Helix and β \beta β Chain distribution , We use it E-H Sequence description Represents the predicted protein fragment . The following table contains E-H Several features described . From the above characteristics , Both basic features and new features can describe the E and H Type of statistics . Because all the above features contain some redundant information and noise . therefore , The selected features are shown in the following table .


The most popular and well-known amino acid signature index is AAindex, It is a digitally indexed website database , Various biology including amino acid residues 、 Physical and chemical properties and characteristics of other forms of protein sequences . meanwhile ,AAindex Contains information on three protein properties :AAindex1、AAindex2 and AAindex3 [27-29]. therefore , The characteristics of several amino acids were used in this study .
边栏推荐
- 一元二次方程到规范场
- Kdevtmpfsi processing of mining virus -- Practice
- [websocket] knowledge points for developing online customer service system meaning of status code returned by websocket
- Kerberoasting without SPN
- 论文阅读 (51):Integration of a Holonic Organizational Control Architecture and Multiobjective...
- Pause update Bulletin - walking Pikachu
- History of storage technology: from tape to hardware liquefaction
- 论文阅读 (49):Big Data Security and Privacy Protection (科普文)
- Wiley-中国科学院文献情报中心开放科学联合研讨会第二讲:开放获取期刊选择及论文投稿...
- Introduction to GTS Academy
猜你喜欢

QML类型:Loader

VNC Viewer方式的远程连接树莓派
iMeta | 南农沈其荣团队发布微生物网络分析和可视化R包ggClusterNet
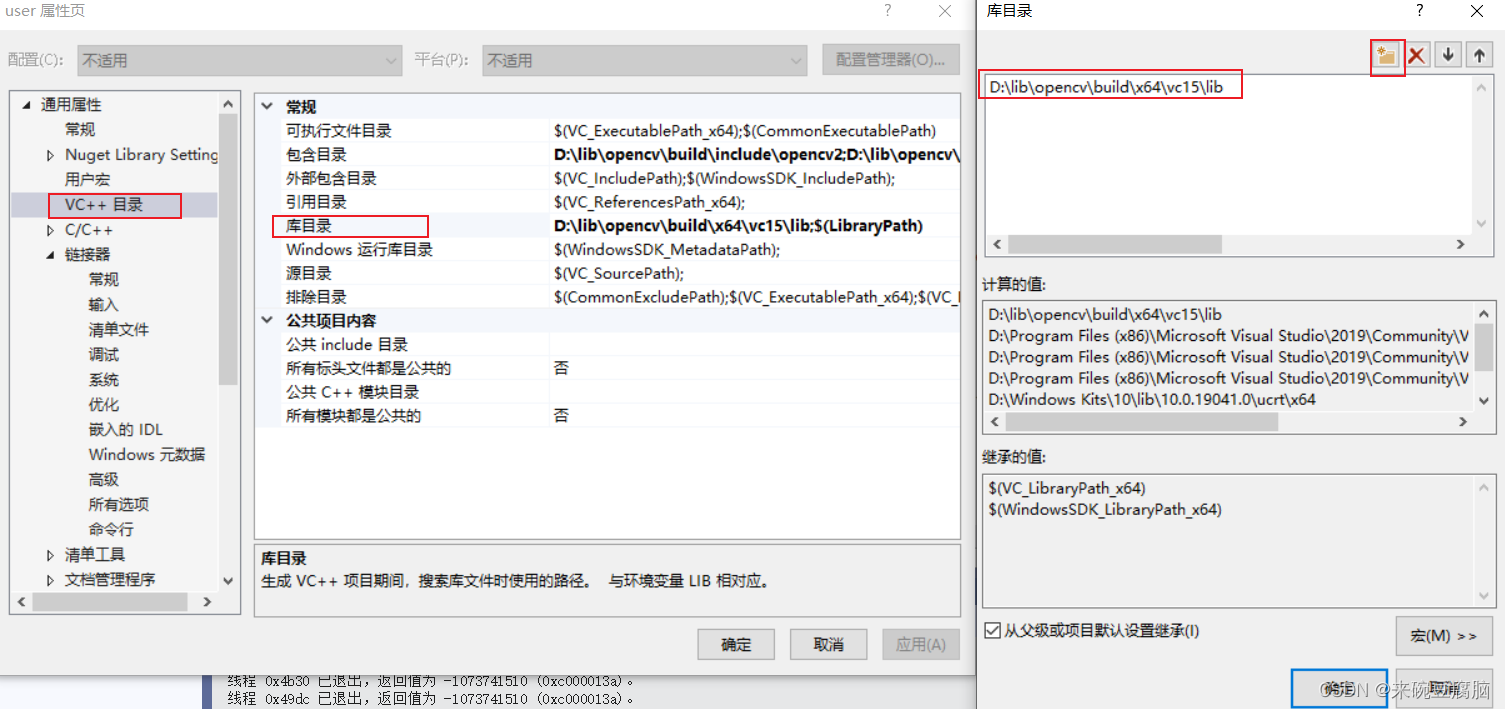
【win10 VS2019 opencv4.6 配置参考】

论文阅读 (58):Research and Implementation of Global Path Planning for Unmanned Surface Vehicle Based...

SimpleDateFormat在多线程环境下存在线程安全问题。

Alien world, real presentation, how does the alien version of Pokemon go achieve?

Paper reading (48):a Library of optimization algorithms for organizational design

论文阅读 (53):Universal Adversarial Perturbations
【Wwise】Wwise嵌入Unity后打包出现没有声音问题
随机推荐
Three functional forms of intelligent switch
Introduction to GTS Academy
Listen attentively and give back sincerely! Pay tribute to the best product people!
Crmeb second open SMS function tutorial
暂停更新公告—行走的皮卡丘
Mobile SSH connection tool
全局组织结构控制之抢滩登陆
VNC Viewer方式的远程连接树莓派
对抗攻击与防御 (2):对抗样本的反制策略
《致敬百年巨匠 , 数藏袖珍书票》
High availability solution practice of mongodb advanced applications (4)
How to solve the problem that the esp8266-01s cannot connect to Huawei routers
How to make validity table
How to design a seckill system?
7、VLAN-Trunk
微信小程序startLocationUpdateBackground()简单实现骑手配送位置
Goframe framework: basic auth Middleware
MySQL transaction submission process
PostgreSQL series articles -- the world's most advanced open source relational database
论文阅读 (50):A Novel Matrix Game with Payoffs of Maxitive Belief Structure