当前位置:网站首页>Hisat2 - stringtie - deseq2 pipeline for bulk RNA seq
Hisat2 - stringtie - deseq2 pipeline for bulk RNA seq
2022-07-03 07:23:00 【Han Jiangang (caas-ucd)】
Software official website :
Hisat2: Manual | HISAT2
StringTie:StringTie
It is recommended to watch the nanny level tutorial :
1. RNA-seq : Hisat2+Stringtie+DESeq2 – Hengnuo Xinzhi
2. RNA-seq use hisat2、stringtie、DESeq2 analysis - Simple books
Basic usage :
1. Build reference genome index
# Extract splice site and exon information
extract_splice_sites.py Mus_musculus.GRCm39.104.gtf > Mus_musculus.ss
extract_exons.py Mus_musculus.GRCm39.104.gtf > Mus_musculus.exon
# Index
# final Mus_musculus.GRCm39_tran Prefix the index file
hisat2-build --ss Mus_musculus.ss --exon Mus_musculus.exon Mus_musculus.GRCm39.dna.primary_assembly.fa \
Mus_musculus.GRCm39_tran
# Time is too long , Greater than 12h, It is recommended to run at night 2. Reference genome alignment
# -x Prefix with index name ,-1,-2 With double ended sequencing files ,-U With single end sequencing files ,-S Output is sam File format ,-p Number of threads
# We directly output the sorted bam file
# --dta Output assembled as transcripts reads,--summary-file Output comparison information
hisat2 -p 10 --dta -x path/to/Mus_musculus.GRCm39_tran
--summary-file test1_summary.txt
-1 1.fastq-data/test1_R1_rep1.fq.gz
-2 1.fastq-data/test1_R2_rep1.fq.gz
-S test1.sam3. samtools For output sam The files are sorted and converted to bam file
# [email protected] by samtools Number of threads for
samtools sort [email protected] 10 -o test1.sorted.bam test.sam4. Transcript assembly
# Assemble transcripts ,-p Number of threads ,-G Refer to the annotation document for assembly ,-l Prefix the output file name
# Single sample run
stringtie -p 10 -G Mus_musculus.GRCm38.102.gtf
-l test1
-o test1.gtf
test1.sorted.bam5. Comment file merge
# establish mergelist.txt file , Indicate the path of the post assembly annotation file
path/to/test1.gtf
path/to/test2.gtf
path/to/test3.gtf
# Merge gtf file
$ stringtie --merge -p 10 -G ./Mus_musculus.GRCm38.102.gtf
-o stringtie_merged.gtf
mergelist.txt6. The transcripts were quantified using the generated annotation file
# Create a new test1 Folder , The quantitative results of transcripts are saved in a folder
mkdir test1/
stringtie -p 10 -e -G ./stringtie_merged.gtf
-o test1/test1.gtf
-A test1/gene_abundances.tsv
test1.sorted.bam
# Generate the sample name under the corresponding folder .gtf and gene_abundances.tsv Two documents of , Corresponding to each sample count Value quantitative results , We need to merge into one file .7. Extract quantitative results of genes
prepDE.py Need one sample_list, The first column is the sample name , The second as gtf File path
# sample_list.txt The contents of the document are as follows
test1 path/to/test1/test1.gtf
test2 path/to/test1/test2.gtf
test3 path/to/test1/test3.gtf
test4 path/to/test1/test4.gtf
# Extract merge count result ,-i For input sample_list
prepDE.py -i sample_list.txt
# Generate gene_count_matrix.csv and transcript_count_matrix.csv file 8. Choose to do : extract FPKM/TPM or coverage result
Need to use stringtie_expression_matrix.pl, The download address is as follows :
rnaseq_tutorial/stringtie_expression_matrix.pl at master · griffithlab/rnaseq_tutorial · GitHub
# extract TPM
$ ./stringtie_expression_matrix.pl --expression_metric=TPM
--result_dirs='test1_rep1,test1_rep2,test2_rep1,test2_rep2'
--transcript_matrix_file=transcript_tpms_all_samples.tsv
--gene_matrix_file=gene_tpms_all_samples.tsv
# extract FPKM
./stringtie_expression_matrix.pl --expression_metric=FPKM
--result_dirs='test1_rep1,test1_rep2,test2_rep1,test2_rep2'
--transcript_matrix_file=transcript_fpkms_all_samples.tsv
--gene_matrix_file=gene_fpkms_all_samples.tsv
# extract coverage
./stringtie_expression_matrix.pl --expression_metric=coverage
--result_dirs='test1_rep1,test1_rep2,test2_rep1,test2_rep2'
--transcript_matrix_file=transcript_coverage_all_samples.tsv
--gene_matrix_file=gene_coverage_all_samples.tsv
# In the current directory, corresponding genes and transcripts will be generated tpm、fpkm、coverage result 9. DESeq2 Difference analysis
# install DESeq2 package
BiocManager::install('DESeq2')
# Load package
library(DESeq2)
# Set work path
setwd('D:rnaseq')
# Read in counts matrix
gene_count_matrix <- read.csv("D:/rnaseq/gene_count_matrix.csv",row.names = 1)
count <- gene_count_matrix[rowSums(gene_count_matrix)>0,]
# Construct phenotype matrix
colData <- data.frame(row.names = colnames(count),
condition = factor(c(rep('control',2),rep('treat',2)),
levels=c('control','treat'))
)
# see
colData
# condition
# test1_rep1 control
# test1_rep2 control
# test2_rep1 treat
# test2_rep2 treat
dds <- DESeqDataSetFromMatrix(countData = count, colData = colData,design = ~ condition)
dds <- DESeq(dds)
res <- results(dds)
diff_res <- as.data.frame(res)
diff_res$gene_name <- rownames(diff_res)
# Output difference results
write.table(diff_res,file = 'DESeq2_diff_results.csv',quote = F,sep = ',',row.names = F,col.names = T)边栏推荐
- SecureCRT password to cancel session recording
- Strategy mode
- 带你全流程,全方位的了解属于测试的软件事故
- Use of other streams
- Laravel Web Framework
- Setting up the development environment of dataworks custom function
- Homology policy / cross domain and cross domain solutions /web security attacks CSRF and XSS
- Advanced API (UDP connection & map set & collection set)
- gstreamer ffmpeg avdec解码数据流向分析
- 2. E-commerce tool cefsharp autojs MySQL Alibaba cloud react C RPA automated script, open source log
猜你喜欢
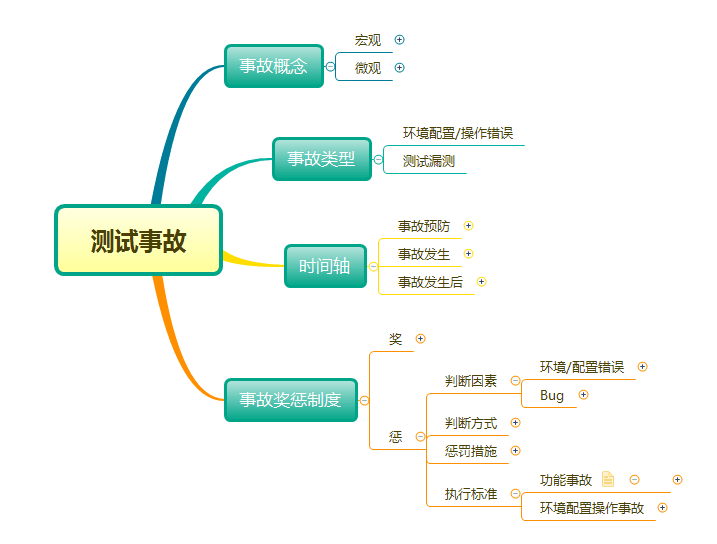
Take you through the whole process and comprehensively understand the software accidents that belong to testing

Use of file class

FileInputStream and fileoutputstream

IO stream system and FileReader, filewriter

IPv4 address

Common methods of file class

Dora (discover offer request recognition) process of obtaining IP address
Interfaces and related concepts
1. E-commerce tool cefsharp autojs MySQL Alibaba cloud react C RPA automated script, open source log

深度学习参数初始化(一)Xavier初始化 含代码
随机推荐
[plus de détails] dernière entrevue complète redis (50)
【已解决】SQLException: Invalid value for getInt() - ‘田鹏‘
SecureCRT password to cancel session recording
2021-07-18
Map interface and method
PHP install the spool extension
Advanced API (byte stream & buffer stream)
FileInputStream and fileoutputstream
IO stream system and FileReader, filewriter
最全SQL与NoSQL优缺点对比
【最詳細】最新最全Redis面試大全(50道)
VMware virtual machine installation
1. E-commerce tool cefsharp autojs MySQL Alibaba cloud react C RPA automated script, open source log
VMWare网络模式-桥接,Host-Only,NAT网络
4everland: the Web3 Developer Center on IPFs has deployed more than 30000 dapps!
Understanding of class
Take you through the whole process and comprehensively understand the software accidents that belong to testing
IPv4 address
[HCAI] learning summary OSI model
Use of framework