当前位置:网站首页>R bioinformatics statistical analysis
R bioinformatics statistical analysis
2022-06-11 03:14:00 【ATU trans】
Perform quantitative RNAseq
use edgeR Estimate the differential expression | use edgeR Estimate the differential expression | Use powsimR Conduct efficacy analysis | Use GRanges Object to find uncommented transcriptional regions | Use bumphunter Look for areas that show high expression from the beginning | Differential peak analysis | Use SVA Estimate the batch effect | Use AllelicImbalance Look for allele specific expression | Draw and render RNAseq data
Use HTS Data search for genetic variation
Use VariantTools Find... In sequence data SNP And insertion missing | Predict the open read frame in the long reference sequence | Use karyoploteR Draw features on the genetic map | Look for alternative transcriptional subtypes | Use VariantAnnotation Select and classify variants | Extract information from genomic regions of interest | Look for and GWAS The phenotype and genotype of | Estimate the number of copies of the site of interest
Search for domains and Motifs of genes and proteins
Looking for a common motif DNA Motif | Use PFAM and bio3d | Look for protein domains | lookup InterPro Domain | Perform multiple comparisons of genes or proteins | Use DECIPHER Align genome length sequences | Machine learning is used to detect new features in proteins | Use bio3d Conduct 3D Structural protein alignment
Use SeqinR Retrieve genome sequence data :
for example , You learned how to learn from NCBI Web site retrieval has NCBI Login number NC_001477 Of DEN-1 Dengue virus genome sequence . To retrieve information with a specific NCBI Added sequence , You can use R function “getncbiseq()”, You first need to copy and paste it into R in :
> getncbiseq <- function(accession)
{
require("seqinr") # this function requires the SeqinR R package
# first find which ACNUC database the accession is stored in:
dbs <- c("genbank","refseq","refseqViruses","bacterial")
numdbs <- length(dbs)
for (i in 1:numdbs)
{
db <- dbs[i]
choosebank(db)
# check if the sequence is in ACNUC database 'db':
resquery <- try(query(".tmpquery", paste("AC=", accession)), silent = TRUE)
if (!(inherits(resquery, "try-error")))
{
queryname <- "query2"
thequery <- paste("AC=",accession,sep="")
query(`queryname`,`thequery`)
# see if a sequence was retrieved:
seq <- getSequence(query2$req[[1]])
closebank()
return(seq)
}
closebank()
}
print(paste("ERROR: accession",accession,"was not found"))
}
Will function getncbiseq() Copy and paste to R In the after , You can use it from NCBI Search sequence in nucleotide database , for example DEN-1 The sequence of dengue virus ( Login number NC_001477):
> dengueseq <- getncbiseq("NC_001477")
Variable dengueseq It is a vector containing nucleotide sequence . Each element of the vector contains a nucleotide of the sequence . therefore , To print out a subsequence of the sequence , We just type in the vector dengueseq The name of , Then enter the square brackets containing these nucleotide indexes . for example , The following command prints DEN-1 The pre genome sequence of dengue virus 50 Nucleotides :
> dengueseq[1:50]
[1] "a" "g" "t" "t" "g" "t" "t" "a" "g" "t" "c" "t" "a" "c" "g" "t" "g" "g" "a"
[20] "c" "c" "g" "a" "c" "a" "a" "g" "a" "a" "c" "a" "g" "t" "t" "t" "c" "g" "a"
[39] "a" "t" "c" "g" "g" "a" "a" "g" "c" "t" "t" "g"
Please note that ,dengueseq[1:50] It's a vector dengueseq The elements of , Its index is 1-50. These elements include DEN-1 Pre dengue virus sequence 50 Nucleotides .
Phylogenetic analysis and visualization
Use ape and treeio Read and write various tree formats | Use ggtree Quickly visualize a tree of many genes | Using tree space to quantify the distance between trees | Use ape Extract and process subtrees | Create a dot chart for alignment Visualization | Use phangorn Rebuild trees from the route
Macrogenomics
Use phyloseq Load hierarchical classification data | Use a meta encoder for sparse counting to correct for sample differences | Use dada2 Read the amplicon data from the original read | Using heat tree to visualize classification abundance in meta encoder | Calculate the sample diversity using the pure element | Split the sequence file into operable taxons
Proteomics from spectroscopy to annotation
Visually represent the original MS data | View proteomic data in the genome browser | Visualize the distribution of peptide hit counts to find thresholds | transformation MS Format to move data between tools | Use protViz Match the spectrum with the peptide for verification | Apply a quality control filter to the spectrum | Identify genomic sites that match peptides
Production release and Web Ready Visualization
Use ridgeplots Visualize multiple distributions | Create a color map for bivariate data | Represent relational data as a network | Use plotly Create interactive Web graphics | Use plotly Build a 3D drawing | Construct a circular genome map of multiple sets of data
Using databases and remote data sources
from BioMart Search for genes and genome annotations | Retrieve and use SNP | Obtain gene ontology information | from SRA/ENA Find experiments and readings in | Perform quality control and filtering on high-throughput sequence reads | Use an external program to complete the reading to reference comparison | Visually read the quality control chart of reference comparison
Useful statistical and machine learning methods
correction p Value to explain multiple assumptions | Generate an analog dataset representing the background | Learn groups in data and use kNN To classify | Use random forest prediction classes | Use SVM Prediction | Learning groups in data without prior information | Use random forests to identify the most important variables in the data | Use PCA Identify the most important variables in the data
Use Tidyverse and Bioconductor Programming
Make basic R The object is clean | Use nested data frames | Written for mutate Function of | Use... Programmatically Bioconductor class | Develop reusable workflows and reports | Use apply Series of functions
Build objects and packages for code reuse
Create simple S3 Object simplifies code | utilize S3 Class | Use S4 The system creates structured and formal objects | A simple way to package code for sharing and reuse | Use devtools Managed from GitHub Code for | Build unit test suites to ensure functionality | Work as you wish | Use Travis To keep code testing and updating
Source code
For details, please refer to - Yatu inter
边栏推荐
- Detailed explanation of unity project optimization (continuous supplement)
- 删除CSDN上传图片的水印
- Demand and Prospect of 3D GIS Industry
- Stringutils string tool class used by FreeMarker to create templates
- What is the difference between a database unique index and a common index?
- B_QuRT_User_Guide(16)
- 数组全全排列
- Application of the remote acquisition IOT gateway of the Bashir trough flowmeter in open channel flow monitoring
- Harris corner detection opencv
- 位置数据融合表3
猜你喜欢

怎样确保消息的可靠性投递?
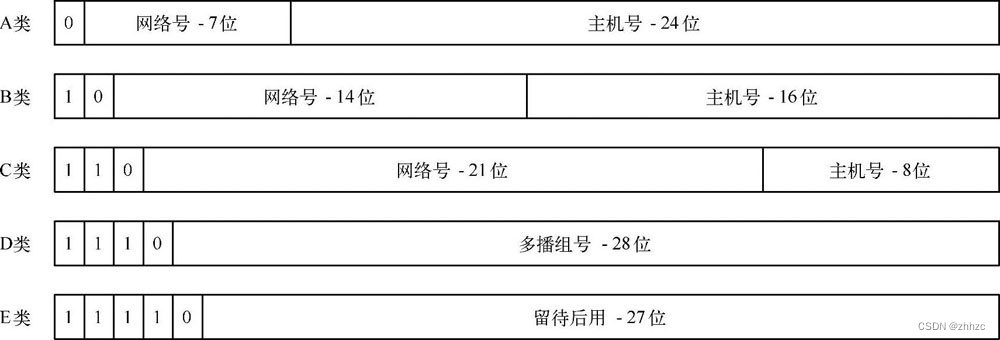
第七章 常用的协议简介(1)
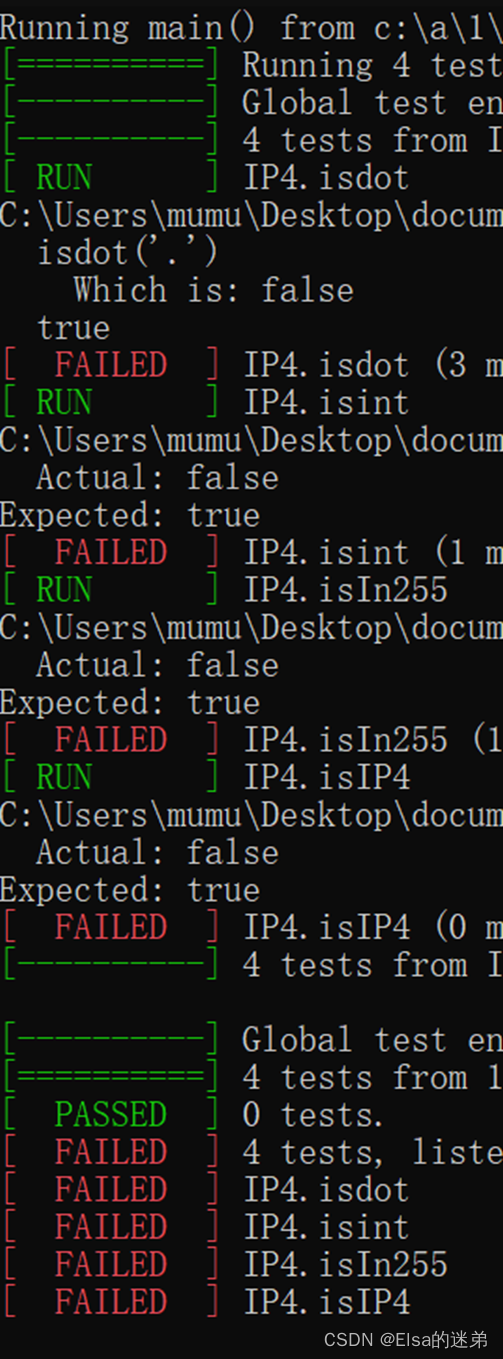
HUST Software Engineering (Experiment 2) -- TDD test driven development experiment.
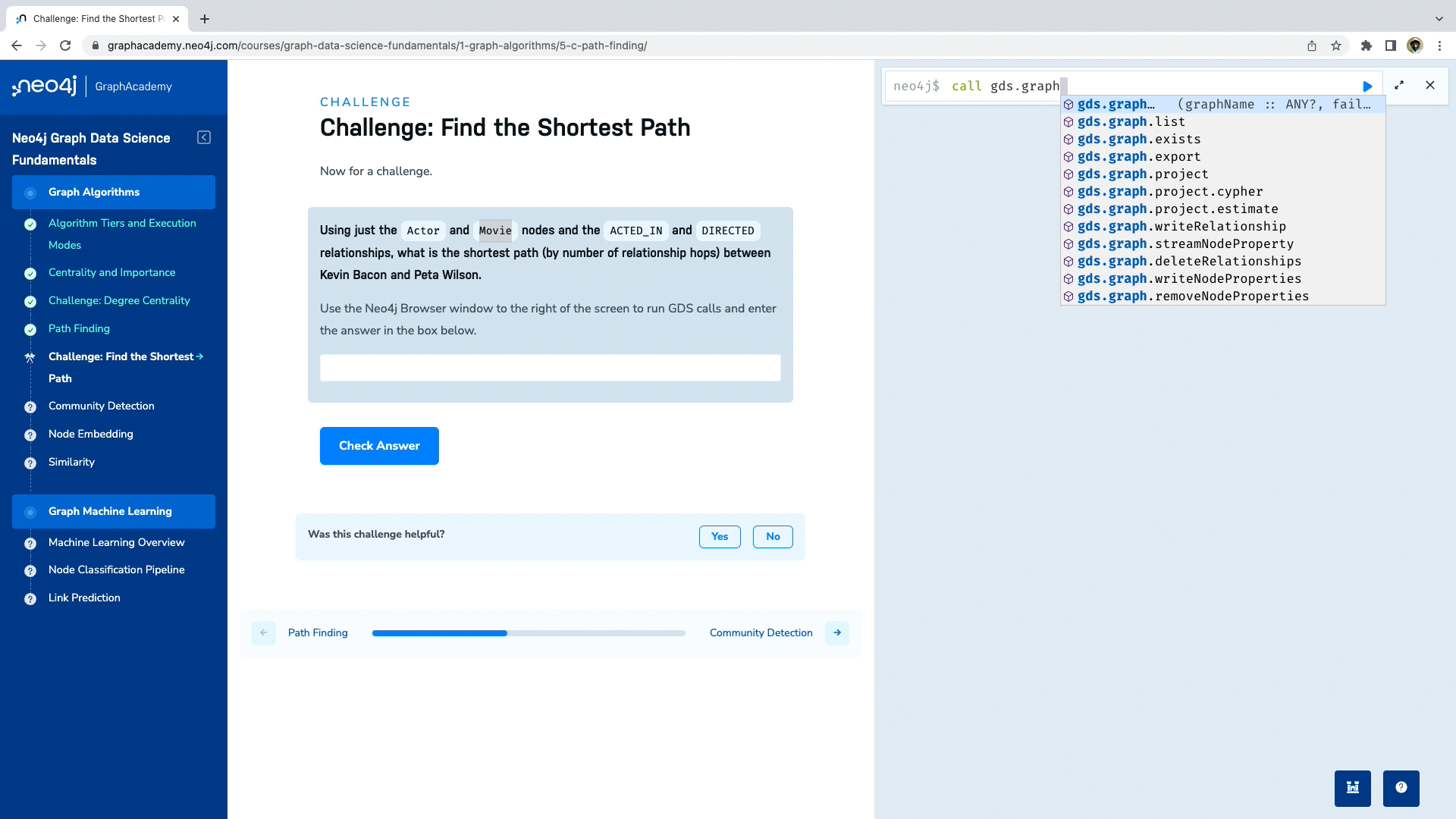
GraphAcademy 課程講解:《Neo4j 圖數據科學基礎》

Help you distinguish GNU, GCC, GCC and G++

Go language advantages and learning Roadmap

ASLR

Basic use of sonarqube platform

计算机视觉(AI)面试大全

How to handle error code 30204-44 when installing office 2016 in win10?
随机推荐
VMware虚拟机IP,网关设置。虚拟机ping不通外网
ArTalk | 如何用最小投入,构建国产超融合进化底座?
Pyqt5:qlineedit control code
The two departments jointly issued the nine provisions on fire safety management of off campus training institutions
File file = new File(“test.txt“)文件路径
Operations on annotation and reflection
Forest v1.5.22 release! Kotlin support
Application of the remote acquisition IOT gateway of the Bashir trough flowmeter in open channel flow monitoring
删除CSDN上传图片的水印
Openjudge noi 1.13 17: text layout
HUST Software Engineering (Experiment 2) -- TDD test driven development experiment.
postgresql源码学习(十八)—— MVCC③-创建(获取)快照
关于玩家身上有个普通Set并发安全的讨论
Arduino uno connected to jq8900-16p voice broadcast module
JS memory leak
CocosCreator原生二次开发的正确姿势
一文搞懂单片机驱动8080LCD
Go quick start of go language (I): the first go program
B_QuRT_User_Guide(17)
TimeHelper