当前位置:网站首页>November 9, 2020 [wgs/gwas] - whole genome analysis (association analysis) process (Part 2)
November 9, 2020 [wgs/gwas] - whole genome analysis (association analysis) process (Part 2)
2022-06-30 07:39:00 【Muyiqing】
2021.07.30 Updated data analysis section : Check me out.
Catalog
One . Abstract
After a period of half a month patchwork R & D testing , The author has finally sorted out a VCF At the beginning GWAS Post analysis process . Of course, I would like to thank a lot of big guys for providing Code help , Reference links are also included in the article . Yes GWAS Friends who are not familiar enough , You can take a look at the one I sorted out before PPT Learning notes 《 Genetic evolution and GWAS Research 》.
Two . SNP Inspection and notes
Use software :snpEff
Installation mode :
miniconda3: conda install snpeff
SnpEff and SnpSift (pcingola.github.io)
Usage flow :
Download the reference genome and annotation file ( Be careful vcf Chromosomes are numbers , Use RefSeq perhaps GenBank Numbering requires uniform chromosome sequence numbering )
java -jar /home/yangxin/miniconda3/pkgs/snpeff-5.0-0/share/snpeff-5.0-0/snpEff.jar build -c /home/yangxin/miniconda3/pkgs/snpeff-5.0-0/share/snpeff-5.0-0/snpEff.config -gtf22 -v GCF_002163495.1_Omyk_1.0_genomic
# Parameter description
#java -jar: Java Run the program in the environment
#-c snpEff.config Profile path
#-gff3 Set the input genome annotation information to gff3 Format , If it is gtf Please change the file to -gtf22
#-v Set the log information output during program operation
final -v Parameters Set the input genome version information , and snpEff.config The information added in the configuration file is consistent
java -jar /home/yangxin/miniconda3/pkgs/snpeff-5.0-0/share/snpeff-5.0-0/snpEff.jar build -c /home/yangxin/miniconda3/pkgs/snpeff-5.0-0/share/snpeff-5.0-0/snpEff.config -gff3 -v GCF_002163495.1_Omyk_1.0_genomic # Build a genome database
Process chromosome numbers ( Optional )
Because the genome I downloaded uses RefSeq Serial number , therefore , I need to change the chromosome number .
awk '{
$1="";print $0}' OFS="\t" AxiomGT1.calls.vcf > col_2.vcf
If your chromosome is not consistent with the database , Then you can manually generate the chromosome sequence file col_chr2.vcf
paste -d '' col_chr2.vcf col_2.vcf >AxiomGT1.vcf # Merge sequence
sed -n l AxiomGT1.vcf # Check vcf Whether the file format is consistent , Note that the separator is “\t”
Be careful :cat、head、less I can't see the problem of the separator
java -jar ~/miniconda3/pkgs/snpeff-5.0-0/share/snpeff-5.0-0/snpEff.jar eff -c ~/miniconda3/pkgs/snpeff-5.0-0/share/snpeff-5.0-0/snpEff.config GCF_002163495.1_Omyk_1.0_genomic AxiomGT1.vcf > AxiomGT1.snp.eff.vcf -csvStats AxiomGT1_positive.csv -stats AxiomGT1_positive.html # Yes snp Annotate
awk -F "|" '{print $1,$2,$4,$5,$8,$9}' OFS="\t" AxiomGT1.snp.eff.vcf > SNV.anno.xls # Generate comment file
# Parameters -F "|" Used to process header information , If the header is normal, this parameter can be omitted
Result display : Notes file form 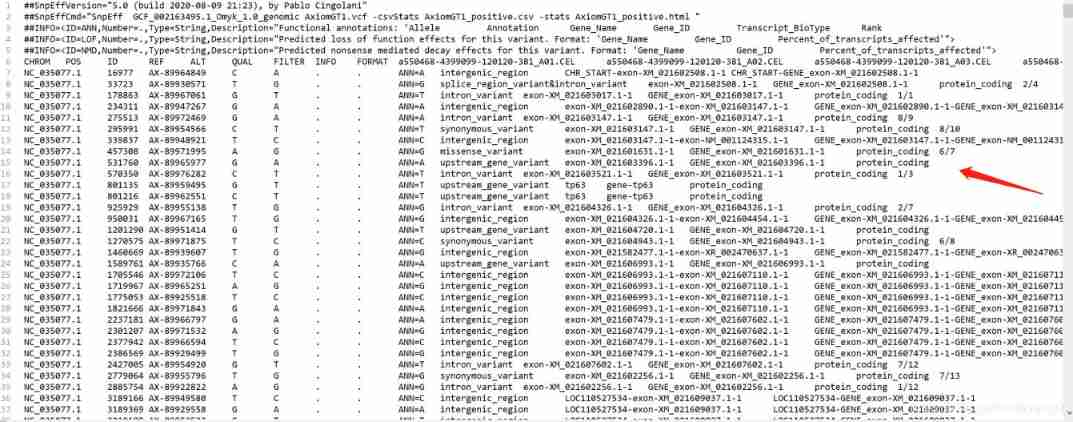
Process reference :SnpEff Usage method
3、 ... and . Groups SNP Filter
Use software :vcftools
Installation mode :
miniconda3: conda install vcftools
Usage flow :
Basic filter parameters Calling rate < 95%, Minor allele count < 3.
vcftools --vcf AxiomGT1.calls.vcf --max-missing 0.95 --mac 3 --recode --recode-INFO-all --out AxiomGT1.calls.filter.vcf
#--max-missing 0.95, The filter calling rate lower than 95% The genotype of
#--mac 3 Filter minor allele count less than 3 Of SNP
vcftools --vcf AxiomGT1.calls.filter.recode.vcf --missing-indv
# Test sample data loss rate , If the loss rate is too high, you need to adjust the parameters .
Result display
Before filtration :
After filtration :
Four . Group stratified analysis
Group stratification refers to the existence of subgroups within a group , The interrelationship among individuals within a subgroup is greater than that among individuals within the entire population Average kinship . Between different subgroups , Some stable alleles have different frequencies , When two subgroups are mixed for association analysis Time analysis , Will lead to false positive results . therefore , Before performing correlation analysis , Be sure to carry out group stratification analysis first .
The group stratification analysis has : Population evolutionary tree analysis and principal component analysis , The results of the two can be mutually verified .
4.1 Population evolutionary tree analysis
There are many software to draw the evolution tree , But after a few tests, I still feel tassel Visual interface drawing is the most convenient , Now let's introduce
Use software :Tassel
Software installation :miniconda3: conda install tassel5.0
Download from the official website :Tassel (bitbucket.io)
Usage flow ( Visual drawing ):
step1: open Tassel
step2: Import and open vcf file 
step3: Create an evolution tree document 
step4: Draw evolution tree 
step5: Adjust the image 
Function introduction :
On the left side, you can add or delete parameters of the input file ;
The lower left corner X,Y± Used to adjust the length and width of the evolutionary tree ;
top left corner File You can output files in various formats ;
On the toolbar Type You can choose the type of evolutionary tree .( The last circle is more beautiful )
Result display

Be careful : because VCF The file in Tassel There is a maximum character limit for association analysis , Just intercept the front 10 Characters , But drawing an evolutionary tree doesn't handle characters . So if you are worried that it is inconvenient to draw the evolution tree, you can handle it in advance .
Tassel The use of reference :https://bitbucket.org/tasseladmin/tassel-5-source/wiki/UserManual
Manual in Chinese (2014):https://wenku.baidu.com/view/4c0cabbe9b6648d7c0c746b2.html
4.2 Group principal component analysis
Use software :Plink
Software installation :miniconda3: conda install Plink
Usage flow :
vcftools --vcf AxiomGT1.calls.vcf --plink --out GT1
# Generate plink Needed ped/map Format
plink --file GT1 --make-bed --out GT1 --chr-set 30
# Convert to bed Format , Non human chromosomes , Use --chr-set Parameter setting chromosome number
Plink --threads 8 --bfile snp --pca 10 --out pca
#--threads Number of threads --bfile Input .bed file --pca The component of the principal component --out Output file name
###########R-PCA###########
mydat<-read.table("pca.eigenvec",as.is = T,header = T,stringsAsFactors = F)
png("PCA.png",width = 7000,height = 7000,pointsize = 160)
Result display 
Tassel,GCTA perhaps EIGENSOFT It can also be analyzed , You can try more .
4.3 Population genetic structure analysis
In this part, I have a special article to introduce the use of error reporting , You can directly jump to the reference :Admixture Population genetic structure analysis
Use software :admixture
Software installation :miniconda3: conda install admixture
Usage flow :
The above is the same as the principal component analysis , Need to use plink take VCF File conversion to bed/bim/fam A few papers
vcftools --vcf AxiomGT1.calls.vcf --plink --out GT1
# Generate plink Needed ped/map Format
plink --file GT1 --make-bed --out GT1 --chr-set 30
# Convert to bed Format , Non human chromosomes , Use --chr-set Parameter setting chromosome number
Admixture Tool handling
for K in 2 3 4 5 6 7 8 9 10; \
do admixture --cv GT1_test.bed $K | tee log${K}.out; done
#2 3 4 5 6 7 8 9 10 The number of group structures divided hapmap3.bed Input file
grep -h CV log*.out
# View best K value ( minimum value ), Best output K Value file or show all K Worth the result
#R mapping
for (k in 2:10){
file_name = paste("GT1_test.",k,".Q",sep="")
result_name = paste(file_name,".PDF",sep="")
pdf(result_name,width=15,height=3)
tbl <- read.table(file_name,header = T)
barplot(t(as.matrix(tbl)), col=rainbow(k),xlab="Individual", ylab="Ancestry", border=NA)
dev.off()
}
Result display :
With k=3,k=8 For example 

5、 ... and . Linkage disequilibrium analysis
2018 year 10 month 15 Japan ,Bioinformatics The magazine published online PopLDdecay Software , Used to analyze the attenuation of linkage disequilibrium (Linkage disequilibrium (LD) decay).PopLDdecay It can be read directly VCF file , Compared to calculating LD frequently-used PLINK Software , This feature simplifies the tedious process of format conversion .
Use software :PopLDdecay
Source download :PopLDdecay-3.41
Usage flow :
If you want to plot by phenotype , We need to build a list, Calculate by phenotype ,list The format is a list of sample names of this phenotype
PopLDdecay -InVCF AxiomGT1.calls.vcf -OutStat RESISTANT -SubPop RESISTANT.list # Parting calculation
gunzip RESiSTANT.stat.gz # Other phenotypic operations are the same
Groups were established according to different phenotypes list, The single line format is stat route , Separator ,stat File prefix 
perl ~/soft/PopLDdecay-3.41/bin/Plot_MultiPop.pl -inList file.list -output Fig
Result display 
Reference link :PopLDdecay: be based on VCF Fast file 、 A tool for efficiently calculating chain disequilibrium
6、 ... and . Select elimination analysis
Population evolutionary selection elimination analysis simply means that a region of the genome is selected to eliminate polymorphism . Selective elimination analysis is the imprint of positive selection on the genome of a species . Compared with the wild ancestors , The regional genetic diversity of cultivated or domesticated species is significantly reduced by selective elimination , This is a typical feature of domesticated areas .
Use software :vcftools
Software installation : A little
6.1 Fst analysis
The fixed coefficient of the group F It reflects the heterozygosity level of population alleles . Fixed coefficient F yes F statistic (Fst) A special case of .Fst The analysis indicates the degree of differentiation of the population , The bigger the value is. , The higher the degree of population differentiation , The more selective .
## For each SNP The variation sites were calculated
vcftools --vcf AxiomGT1.calls.vcf --weir-fst-pop 1_population.txt --weir-fst-pop 2_population.txt --out Fst_AximoGT1
## Calculate in steps
vcftools --vcf test.vcf --weir-fst-pop 1_population.txt --weir-fst-pop 2_population.txt --out p_1_2_bin --fst-window-size 500000 --fst-window-step 50000
#test.vcf yes SNP calling Generated after filtering vcf file ;
#p_1_2_3 Generate the result prefix
#1_population.txt It's a document , Include all individuals in group one , Usually one individual per line . The individual name should be the same as vcf The name of corresponds to .
#2_population.txt It includes all the individuals in group 2 .
# The calculation window is 500kb, And the step size is 50kb ( It can be adjusted according to your needs ). We can also calculate only for each point Fst, Remove the parameters (--fst-window-size 500000 --fst-window-step 50000) that will do .
#R-manhattan mapping
library(qq)
data<-read.table("Fst_AxiomGT1.weir.fst",header=T)
pdf(file="Fst_manhattan.pdf",width=20,height=8)
png(file="Fst_manhattan.png",width=2400,height=960)
#sc3 = subset(data,CHROM=="1") Select any chromosome for analysis
manhattan(data,chr="CHROM",bp="POS",p="WEIR_AND_COCKERHAM_FST",logp=F,main="Manhattan Plot",col = c("#8DA0CB","#E78AC3","#A6D854","#FFD92F","#E5C494","#66C2A5","#FC8D62"),annotatePval = 0.01)
dev.off()
Result display

6.2 θπ analysis
π To analyze base polymorphism , The lower the polymorphism , The more selective .
vcftools --vcf AxiomGT1.calls.vcf --keep RESISTANT.list --recode --out AxiomGT1.RESISTANT
vcftools --vcf AxiomGT1.calls.vcf --keep SUSCEPTIBLE.list --recode --out AxiomGT1.SUSCEPTIBLE_500K
# Classify the two phenotypes
vcftools --vcf AxiomGT1.RESISTANT.recode.vcf --window-pi 500000 --window-pi-step 500000 --out AxiomGT1.RESISTANT_500K
# With 500K Calculate the step size separately pi value
paste -d '\t' RESISTANT_typename.list AxiomGT1.RESISTANT_500K.windowed.pi > type_RESISTANT_500K.windowed.pi
# Create phenotype labels separately list, to windowed.pi The document is labeled with phenotype
#R mapping
library(ggplot2)
data<-read.table("allsample_500K.windowed.pi",header=T)
for (i in 1:29){
#result_name = paste("chr",i,".pdf",sep="")
#pdf(result_name,width=15,height=3)
result_name = paste("chr",i,".png",sep="")
png(result_name,width=1500,height=300)
chrom = subset(data,CHROM==i)
xlab = paste("Chromosome",i,"(MB)",sep="")
p <- ggplot(chrom,aes(x=BIN_END/1000000,y=PI,group=Phenotype,color=Phenotype,shape=Phenotype)) + geom_line(size=0.5) + xlab(xlab)+ ylab("Pi") + theme_bw()
print(p)
dev.off()
}
Result display 
7、 ... and . Genome wide association analysis
Genome wide association analysis (Genome-wide association study,GWAS) It is a common genetic variation in the whole genome ( Single nucleotide polymorphisms and copy numbers ) The method of gene association analysis , This method takes the natural population as the research object , With a gene that remains after long-term recombination ( site ) Linkage disequilibrium (linkage disequilibrium, LD) Based on , Combining phenotypic diversity of target traits with genes ( Or marker sites ) The polymorphisms of , It can be directly identified to be closely related to phenotypic variation A gene or marker site that is related and has a specific function . use GWAS Technology is studied on a genome-wide scale , It can locate multiple traits at one time , It is suitable for locating the correlation interval of traits 、 Functional gene research 、 Research on the development of trait breeding markers .
There are many software available for analysis , Such as GEMMA、Plink perhaps Tassel. The analysis method is also divided into general linear model and mixed linear model . Here we use GEMMA Analysis of cases .
7.1 Trait association analysis
Use software :GEMMA
Source download :Releases · genetics-statistics/GEMMA · GitHub
Usage flow :
vcftools --vcf AxiomGT1.calls.vcf --plink --out GT1
# Generate plink Needed ped/map Format
plink --file GT1 --make-bed --out GT1 --chr-set 30
# Convert to bed Format , Non human chromosomes , Use --chr-set Parameter setting chromosome number
~/soft/gemma-0.98.1-linux-static -bfile GT1 -gk 2 -p p.txt
~/soft/gemma-0.98.1-linux-static -bfile GT1 -k output/result.sXX.txt -lmm 1 -p p.txt
#-bfile Read plink Binary file (bed\bim\fam)
#-gk 2 Standardized method of calculation G matrix
#-p Read phenotype data ( Generate... Manually , It can't be omitted )
The following uses the name qqman Of R Package drawing Manhattan map and qq chart , Download and install in advance
library(qqman) # Load the package
data <- read.table("result.assoc.txt",header = TRUE) # Reading data
data1 <- data[,c(1,2,3,12)] # Intercept columns according to rules
data2 <- na.omit(data1) # Delete contains NA Entire line of
par(cex=0.8) # Set point size
#color_set <- rainbow(9)
The previous analysis steps are generic , Later, all chromosomes and single chromosomes were analyzed .
All chromosomes manhattan chart
#pdf(file="GWAS_manhattan.pdf", width=20, height=8)
png(file="GWAS_manhattan.png",width=2000, height=800)
manhattan(data2,chr="chr",snp="rs",bp="ps",p="p_wald",main="Manhattan Plot",col = c("#8DA0CB","#E78AC3","#A6D854","#FFD92F","#E5C494","#66C2A5","#FC8D62"),annotatePval = 0.01) #suggestiveline = FALSE More significant
dev.off() # Save the picture
Result display 
A single chromosome manhattan chart
data <- read.table("result.assoc.txt",header = TRUE) # Reading data
data1 <- data[,c(1,2,3,12)] # Intercept columns according to rules
data2 <- na.omit(data1) # Delete contains NA Entire line of
par(cex=0.8) # Set point size
for (i in 1:29){
result_name = paste("chr",i,".pdf",sep="")
pdf(result_name,width=15,height=6)
#result_name = paste("chr",i,".png",sep="")
#png(result_name,width=900,height=450)
chrom = subset(data2,chr==i)
main_title = paste("Chromosome",i,"(MB)",sep="")
manhattan(chrom,chr="chr",snp="rs",bp="ps",p="p_wald",main=main_title,col = c("#8DA0CB","#E78AC3","#A6D854","#FFD92F","#E5C494","#66C2A5","#FC8D62"),suggestiveline = FALSE, annotatePval = 0.01)
#suggestiveline = FALSE Remove the difference significance line
dev.off()
}
Result display 
draw qq chart
pdf(file=“qqplot.pdf”, width=8, height=8)
png(file=“qqplot.png”)
qq(data2$p_wald) # draw qq chart
dev.off()
Result display 
7.2 Multiple hypothesis test correction
Yes GEMMA Got result.assoc.txt Cut the document
awk '{print $2,$1,$3,$12}' result.assoc.txt >GWAS.adjust.xls
stay excel According to the P Value to find the correction value , The specific operations are as follows: p Sort values in ascending order , The correction value formula is ,FDR=p*m/k ,m by snp total ,k For the current snp ranking .
Result display 
7.3 Notes on gene function of target trait related regions
adopt gwas4d Make online comments ( Then I will write a separate article to explain )
Result display 
Reference link : The root is red and the seedling is positive GWAS Analysis software :GEMMA_ Deng Fei ---- Analysis of breeding data -CSDN Blog
8、 ... and . Conclusion
Compared with the process of collecting references for half a month , This article has made some supplements and improvements . However , There is still a lot of room for optimization in all aspects , therefore , It's just a 1.0 edition , If there is a problem with the process , Or offer better advice , When the QR code expires, you can add VX:bbplayer2021 , remarks Apply to join the student information exchange group .
Nine . Reference link
SnpEff Usage method :https://www.jianshu.com/p/a6e46d0c07ee
Tassel The use of reference :https://bitbucket.org/tasseladmin/tassel-5-source/wiki/UserManual
Tassel Manual in Chinese (2014):https://wenku.baidu.com/view/4c0cabbe9b6648d7c0c746b2.html
PopLDdecay: be based on VCF Fast file 、 A tool for efficiently calculating chain disequilibrium :http://wap.sciencenet.cn/blog-656335-1168505.html
The root is red and the seedling is positive GWAS Analysis software :GEMMA_ Deng Fei ---- Analysis of breeding data -CSDN Blog https://blog.csdn.net/yijiaobani/article/details/106215223
边栏推荐
- C language operators
- Implementation of double linked list in C language
- Final review -php learning notes 9-php session control
- DXP shortcut key
- Calculate Euler angle according to rotation matrix R yaw, pitch, roll source code
- Shell command, how much do you know?
- MCU essay
- 期末複習-PHP學習筆記5-PHP數組
- 2022 Research Report on China's intelligent fiscal and tax Market: accurate positioning, integration and diversity
- Virtual machine VMware: due to vcruntime140 not found_ 1.dll, unable to continue code execution
猜你喜欢

Final review -php learning notes 7-php and web page interaction

Basic knowledge of compiling learning records

Common sorting methods

Network security and data in 2021: collection of new compliance review articles (215 pages)

冰冰学习笔记:快速排序

Adjacency matrix representation of weighted undirected graph (implemented in C language)

Examen final - notes d'apprentissage PHP 3 - Déclaration de contrôle du processus PHP

Armv8 (coretex-a53) debugging based on openocd and ft2232h

Personal blog one article multi post tutorial - basic usage of openwriter management tool

C51 minimum system board infrared remote control LED light on and off
随机推荐
DXP software uses shortcut keys
Graphic explanation pads update PCB design basic operation
Distance from point to line
Private method of single test calling object
Stepper motor
LabVIEW program code update is slow
Tencent and Fudan University "2021-2022 yuan universe report" with 102 yuan universe collections
Final review -php learning notes 6- string processing
Binary tree related operations (based on recursion, implemented in C language)
Assembly learning register
Final review -php learning notes 3-php process control statement
C language implementation of chain stack (without leading node)
C language implementation sequence stack
Xiashuo think tank: 50 planet updates reported today (including the global architects Summit Series)
DS1302 digital tube clock
Cadence physical library lef file syntax learning [continuous update]
期末复习-PHP学习笔记1
RT thread kernel application development message queue experiment
November 22, 2021 [reading notes] - bioinformatics and functional genomics (Chapter 5, section 4, hidden Markov model)
6月底了,可以开始做准备了,不然这么赚钱的行业就没你的份了