当前位置:网站首页>Multiomics single cell data integration and regulatory reasoning based on graph linked embedding
Multiomics single cell data integration and regulatory reasoning based on graph linked embedding
2022-06-28 22:24:00 【tzc_ fly】

Front content
Single cell multiomics
For single cell multiomics (Single Cell Multi-Omic), Single cell sequencing counting has been developed so far , From the beginning scRNA-seq,scDNA-seq, Up to now scATAC-seq, Single cell methylation sequencing , Single cell proteome sequencing and other sequencing technologies , So that we have a better understanding of embryonic development , Brain Neuroscience , And cancer and so on , Really start from the cellular level , So that we can carry out research at the functional level of cells , So that we can better understand how genes affect individual traits by affecting the phenotype of cell subpopulations . For Reproductive Medicine , Precision medicine is of great significance .
However , As far as the vast majority of current sequencing technologies are concerned , After collecting data from a batch of cells, only one dimension of information can be obtained , such as , In the use of scRNA-seq When , We can only get the gene expression data of these cells , But I don't know its DNA Methylation modification or proteomic data . But often , Get a cell ( Or a subpopulation of cells ) Multiple omics information is important , This means that we can establish links between different omics data , Better depict the functions of cells and their internal regulatory processes . Combine several of these dimensions of data into a multinomial analysis of the same single cell , It will have an important impact in the fields of basic biology and biomedicine .
Multi source heterogeneous data
Multi source heterogeneous data , That is, data from different sources with different feature types but describing the same object , Concept and multimodality of multi-source heterogeneous data (multi-modal) similar , But multi-source heterogeneous data includes more data types , In the field of information , Mode can be understood as the existence of data format , For example, text , Audio , Images , Video and other formats . When there are multiple modes at the same time, it is called multimodal , For example, as a multimedia video, it can be decomposed into a variety of single-mode data , Such as images , Voice and text .
In fact, the integration of single cell multiomics data is very similar to a multi-source heterogeneous data fusion problem , such as , When we have hematopoietic stem cells scRNA-seq And scATAC-seq Data with two different characteristics , How to integrate these two types of data according to their potential cell subgroup types , For example, we integrate and cluster these two types of data , Suppose they belong to the same T Cell subsets , Chromatin open information belonging to this subgroup (scATAC-seq) And gene expression information (scRNA-seq) Will be divided together .
But it is unreasonable to directly use the characteristics of these two data , Because the characteristics of these two types of data are inconsistent , Therefore, we need to learn technology through certain representations , Get all the samples in the same space ( manifold ) In the vector , Define each cell again ( sample ) The distance between them can be used for subsequent clustering integration .
Single cell data integration is a multi-source data fusion problem , There are many sources , It means more than one experiments Or technology ( batch ), When these experiments The resulting data has the same characteristics , That is, isomorphic or similar data , That is, the well-known problem of removing batch effect , For example, for different sources ( Sequencing platform , laboratory ) The resulting gene expression profile data , because ” Different sources ” Cause noise , Therefore, it is necessary to perform batch correction on the expression profile data .
Compared with multi-source isomorphic data fusion ( Remove the batch effect ), Multi source heterogeneous data integration is a more extensive and difficult task . An important problem is how to embed different features of two datasets into the same manifold space , It makes it possible to measure , The corresponding cell to cell distance .
integration An important assumption of is : Even from different sources , Characteristic of different types dataset, Their potential cell subpopulations are roughly the same , So these dataset( At least part of the information ) It is possible to make connections , Because sharing information about the same object . But at the same time integration And hope to ensure that every dataset Truly unique information can also be preserved , For example dataset A There is a certain cell type that does not belong to dataset B, So in integration, After clustering , These belong only to dataset A Cell types in should not be associated with dataset B Of the cells have any overlap , Otherwise, it is over correction (over-correct).
Integration of homogeneous data and heterogeneous data , All hope :
- As close as possible to cells from the same cell subpopulation in different data sets , That is, they are as close as possible in the manifold space we want ;
- As far as possible, specific cell subpopulation information in different data sets should be retained ;
Pay attention to Paper reading notes - utilize Scanorama Efficient integration of heterogeneous single cell transcriptome Heterogeneity in , It is better to refer to the multi-source isomorphic data described in this chapter . A broad sense , The existing scRNA-seq The integration method can also integrate multi omics data , Because we can assume that the dimension of heterogeneous data sets is reduced to embedding The representation of is the same feature space , Then we can use isomorphic integration to integrate these embedding data .
Abstract
Despite the emergence of experimental methods to simultaneously measure multiple omics modes in a single cell , But most single-cell datasets contain only one mode . A major obstacle to integrating omics data from multiple modalities is , Different omics data usually have different feature spaces . ad locum , We put forward a proposal called GLUE(graph-linked unified embedding) Computing framework of , The framework bridges the modal gap by explicitly modeling interactions across omics . The benchmark test of the system shows that ,GLUE For single-cell heterogeneous multiomics data , More accurate than the most advanced work 、 More robust and scalable . We will GLUE Applied to a variety of challenging tasks , Including the integration of three groups 、 Regulatory reasoning and the construction of a multiomic human cell map of millions of cells ,GLUE Comments that can correct previous data errors .GLUE Modular design , Flexible expansion and enhancement for new analysis tasks .
Main
Recent technological advances in single cell sequencing have enabled us to mine maps through multiomics data , For example, chromatin accessibility chromatin accessibility(scATAC-seq),DNA Methylation (snmC-seq,sci-MET) And single cell transcriptome single cell transcriptome(scRNA-seq), It provides an opportunity to reveal the functions of different cell types . Although recently, there have been methods to analyze multiomics data at the same time , But different omics are usually measured independently , And produce mismatched data , This requires us to develop efficient multi group integration technology .
In calculation , Integrate unpaired multiomics data ( Also known as diagonal integration ) One of the main obstacles is that different omics have different characteristic spaces ( for example ,scATAC-seq The accessible chromatin regions in are related to scRNA-seq Genes in ). The concise method is to transform multimodal data into a common feature space based on prior knowledge , Then apply the data integration method of single omics . This kind of clear “ Feature conversion ” It's easy , But it often leads to information loss . The algorithm based on coupling matrix decomposition avoids explicit transformation , But it can hardly process more than two omics data . Another option is to match cell data from different omics by nonlinear manifold alignment , This completely eliminates the need for prior knowledge , And it can reduce the loss of information between modes in theory ; However , This technique is mainly applied to data sets with a limited number of cell types and a relatively small number of cells .
The growing amount of data is another serious challenge . Recently developed sequencing techniques can usually obtain millions of cell scale data sets , The current integration method is only applicable to data sets with smaller data volume . In order to keep up with the growth of data volume , The design of the integration method should consider multi-scale .
Here it is , We proposed GLUE(graph-linked unified embedding), This is a modular framework , It is used to integrate unpaired single cell multiomics data and realize regulatory reasoning at the same time . By explicitly modeling the interactions between the various omics ,GLUE The gap between the specific feature spaces of different omics is bridged in a biological intuitive way . System benchmarks and case studies show that ,GLUE For single cell multiomics data integration is accurate 、 Reliable and scalable . Besides ,GLUE Designed as a general framework , Allow easy expansion in a modular way .
Results

- chart 1:GLUE The architecture of . The unpaired three omics data were recorded as X 1 ∈ R N 1 × ∣ V 1 ∣ , X 2 ∈ R N 2 × ∣ V 2 ∣ , X 3 ∈ R N 3 × ∣ V 3 ∣ \textbf{X}_{1}\in R^{N_{1}\times |V_{1}|},\textbf{X}_{2}\in R^{N_{2}\times |V_{2}|},\textbf{X}_{3}\in R^{N_{3}\times |V_{3}|} X1∈RN1×∣V1∣,X2∈RN2×∣V2∣,X3∈RN3×∣V3∣, among , N 1 , N 2 , N 3 N_{1},N_{2},N_{3} N1,N2,N3 Is the number of cells , V 1 , V 2 , V 3 V_{1},V_{2},V_{3} V1,V2,V3 It is the feature set of each omics ,GLUE Low dimensional learning from each omics data using a omics specific variational self encoder embedding U 1 , U 2 , U 3 \textbf{U}_{1},\textbf{U}_{2},\textbf{U}_{3} U1,U2,U3. Dimensions of raw data and VAE The resulting distribution can remain different across different omics , but embedding Dimensions m m m It should be shared . To link omics specific data spaces ,GLUE With guidance graph G = ( V , E ) G=(V,E) G=(V,E) The form of the takes advantage of prior knowledge , Where nodes V = V 1 ∪ V 2 ∪ V 3 V=V_{1}\cup V_{2}\cup V_{3} V=V1∪V2∪V3 Is characteristic of different omics . Graph variational self encoder is based on prior knowledge guidance graph(the prior knowledge-based guidance graph) Learning characteristics of the group embedding V = ( V 1 T , V 2 T , V 3 T ) T \textbf{V}=(\textbf{V}^{T}_{1},\textbf{V}^{T}_{2},\textbf{V}^{T}_{3})^{T} V=(V1T,V2T,V3T)T, Then use this in the data decoder guidance graph, By interacting with cells embedding To reconstruct the data of omics by inner product , And effectively link the specific data space of omics , To ensure consistency embedding Direction . Last , Using the omics discriminator D D D Aligning cells of different omics through antagonistic learning embedding. ϕ 1 , ϕ 2 , ϕ 3 , ϕ G \phi_{1},\phi_{2},\phi_{3},\phi_{G} ϕ1,ϕ2,ϕ3,ϕG Represents the learnable parameters in the data encoder and the graph encoder . θ 1 , θ 2 , θ 3 , θ G \theta_{1},\theta_{2},\theta_{3},\theta_{G} θ1,θ2,θ3,θG Represents the learnable parameters in the data decoder and the graph decoder . ψ ψ ψ Represents the learnable parameter in the omics discriminator .
- Because it is a graph VAE, Therefore, the output control chart can be used as the result of control reasoning .
Inspired by previous research , We modeled the cell state as a low dimensional cell embedding via variational self coder learning . In view of their inherent differences in biological properties and analytical techniques , Each omics layer is equipped with a separate self encoder , The encoder customizes the probability model for the feature space specific to the omics layer .
Using previous biological knowledge , We recommend using knowledge-based graphs (guidance graph), Define the regulatory role of features between the modeling cross organizational levels , To link feature spaces specific to the omics layer ; The vertices in the graph correspond to the features of different omics layers , Edges represent the regulatory role between features . for example , When integration scRNA-seq and scATAC-seq Data time , The apex is the gene (gene) And accessible chromatin regions ( namely ATAC peak), An accessible region can be linked to its putative downstream gene . then , In the figure of the encoder feature embedding Under the guidance of , The multimodal alignment is carried out in the form of iterative optimization .

- chart 2: Consolidated performance .
- a: The score of biological conservatism and the score of omics integration of different integration methods ;
- b: Comprehensive scores of different methods ;
- c: Single cell level alignment error of different methods ;
- d: The performance of integration methods that depend on prior feature relations under different prior knowledge damage rates FOSCTTM Increasing trend ;
- e: Different integration methods on sub sample datasets of different sizes FOSCTTM value ;
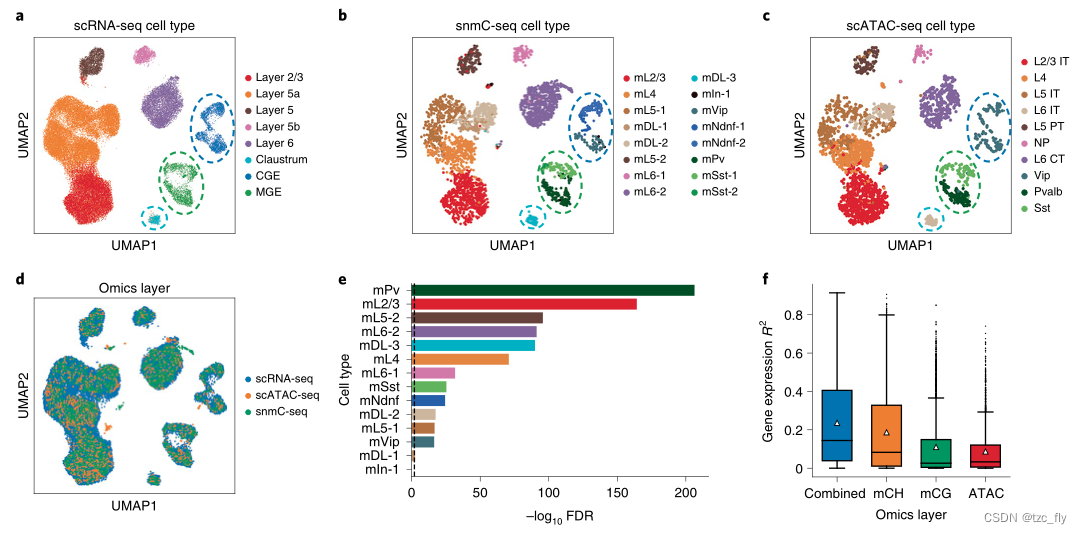
- chart 3: The integration of three groups in mouse cortex . Colored by primitive cell types scRNA-seq(a)、snmC-seq(b) and scATAC-seq(c) Of embedding UMAP visualization . And “mPv” and “mSst” The aligned cells are highlighted with green circles . And “mNdnf” and “mVip” Aligned cells are highlighted with dark blue circles . And “mDL-3” The aligned cells are highlighted with light blue circles .
- d: All integrated cells embedding Of UMAP visualization , Colored by the omics layer .
- e: The significance of marker gene overlap for each cell type in all three omics layers .
边栏推荐
- Laravel文档阅读笔记-Adding a Markdown editor to Laravel
- #yyds干货盘点# 解决剑指offer: 连续子数组的最大和(二)
- 代码复查
- 【HackTheBox】dancing(SMB)
- 场景化接口开发利器,金蝶云苍穹新版OpenAPI引擎来了!
- How many stages did the development and evolution of data analysis go through?
- ROS 2 Humble Hawksbill 之 f1tenth gym
- F1tenth gym of ROS 2 humble hawksbill
- Use of axurer9 master
- IPv6 comprehensive experiment
猜你喜欢

什么是低代码开发?
Laravel文档阅读笔记-Adding a Markdown editor to Laravel

IC Nansha|AMD高级副总裁、大中华区总裁潘晓明:制程、架构、平台优化突破计算边界

爱数SMART 2022峰会开启,分享数据战略与建设数据驱动型组织方法论

Rosdep update using fishros to solve ros1/ros2 problems 2022

Detailed explanation of Zadig's self-test and joint debugging sub environment for developers

Zadig + SonarQube,为开发过程安全保驾

Appium automated test Jiugongge unlock

Architecture design of e-commerce secsha system

Deploy grafana to realize graphical monitoring
随机推荐
In one sentence, I will tell you the meaning of select 1, 2 and 3 in SQL injection, and explain the meaning of each part of SQL injection in detail
Deploy grafana to realize graphical monitoring
穿越过后,她说多元宇宙真的存在
2022-06-28:以下golang代码输出什么?A:true;B:false;C:panic;D:编译失败。 package main import “fmt“ func main() {
Oracle删除归档日志及添加定时任务
Dart的类扩展、可选类型扩展
什么是低代码开发?
职业问诊 | 面试中被问到意向薪资时,该怎么回答?
Zadig + 洞态 IAST:让安全溶于持续交付
稳!上千微服务如何快速接入 Zadig(Helm Chart 篇)
论文解读(DCN)《Towards K-means-friendly Spaces: Simultaneous Deep Learning and Clustering》
Adding a markdown editor to lavel
Pat 1054 the dominiant color (20 points)
BOE was brilliant for the Winter Olympics, but revealed another Chinese technology enterprise dominating the world
How to make beautiful pictures
【网关开发】ngx嵌套lua时处理CIDR表示的IP地址段
Description détaillée du schéma technique du sous - environnement syntonique auto - test de Zadig pour les développeurs
JS array common methods
Linux安装mysql5.7(CentOS7.6) 教程
torch.nn.Transformer导入失败