当前位置:网站首页>The combination of deep learning model and wet experiment is expected to be used for metabolic flux analysis
The combination of deep learning model and wet experiment is expected to be used for metabolic flux analysis
2022-07-05 08:49:00 【Python code doctor】
background
On the road of protein structure transformation , Need powerful computer programming assistance , In the field of deep learning , Found a powerful tool , So study how to combine with wet experiment .
Tools
DeeplearningApproach Anconda python And various dependent packages Computer Wet experimental data
Realize the idea
1. First , Good configuration DeeplearningApproach, How to configure see another article 《 Deep learning predicts enzyme activity parameters, improves enzyme constraint model construction, and builds ab initio environment 》.
2. Use the mutation sites with existing data to predict , For example, I want to predict the sequence of a protein L59,W60,Y153,R416 Site mutation to alanine (A), So I need to configure my tsv file , I wrote a script for the mutation here , The code is as follows :
import copy
def sort(a,b,c,d):
for n,i in enumerate(a):
if n+1 == b: # Determine whether it is the site I want
d = a[n] # Assign this site of the original sequence to d
a = copy.deepcopy(a) # Here you need a deep copy , Otherwise it will change WT Sequence , Of course, if it is the same site, it can be predicted as others 19 Amino acids can be used without
a[n] = c # This site of the original sequence is replaced by the site I need ,
i = a[n] # Extract the changed amino acids
break
else:
a[n] = i
a = ''.join(a)
# print(' Has already put the first %d Amino acids %s Change it to %s, The output sequence is as follows :%s'%((n+1),d,i,a))
print(' Enter the substrate name '+' Enter the chemical formula of the substrate '+a) # The difference between a plus sign and a comma
def main():
# Manual input
# while True:
# a = list(input(' Please enter the amino acid sequence :'))
# b = int(input(' Please select the amino acid site to be modified :'))
# c = input(' Please select the single letter abbreviation of amino acid to be changed :')
# sort(a,b,c,d=None)
aa_list = ['A','G','V','L','I','P','F','Y','W','S','T','C','M','N','Q','D','E','K','R','H']
pro = ' Input amino acid sequence '
pro_list = []
for p in pro:
pro_list.append(p)
for i in aa_list:
sort(pro_list,231,i,d=None)
if __name__ == '__main__':
main()
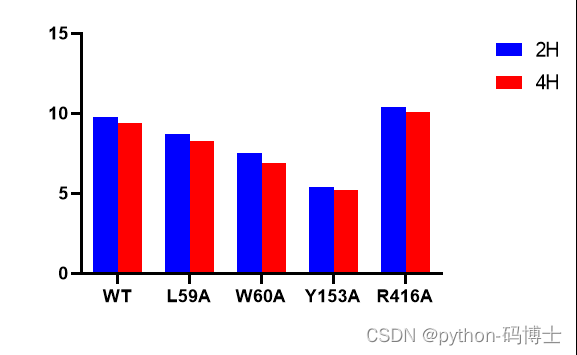
3. By extraction WT,L59,W60,Y153,R416 Of Kcat Value to draw a column chart ( Of course, it's OK to look without drawing , Just a few data ), Compare with the existing experimental data :


Deep excavation
1) How to use it DeeplearningApproach characteristic ?
Make one tsv library , This library contains unit point mutations and double site mutations of all amino acids near the active site of the protein , And WT Of Kcat It's worth comparing , Screen beneficial mutations for experiments .
2) Can you make DeeplearningApproach More suitable for a certain exact experiment , Make it Kcat The value is more accurate , And then use it Kcat Do metabolic flux analysis , Maybe we can start from two aspects :
- Adjust the parameters , To optimize the fitting / Under fitting phenomenon , Do some super parameter optimization , Make it more appropriate to the existing experimental results .
- Using a large amount of experimental data ( At least one million ) Retraining the model , Get a model with strong specificity ( It is almost impossible to achieve , Just a guess )
Be careful
The experimental data are fictitious , To introduce the method .
边栏推荐
猜你喜欢
Count of C # LINQ source code analysis
Halcon: check of blob analysis_ Blister capsule detection

Halcon blob analysis (ball.hdev)
Arduino+a4988 control stepper motor

微信H5公众号获取openid爬坑记

Halcon clolor_ pieces. Hedv: classifier_ Color recognition
Mathematical modeling: factor analysis
Install the CPU version of tensorflow+cuda+cudnn (ultra detailed)

猜谜语啦(4)
Run menu analysis
随机推荐
轮子1:QCustomPlot初始化模板
Pytorch entry record
Ros-10 roslaunch summary
整形的分类:short in long longlong
资源变现小程序添加折扣充值和折扣影票插件
12、动态链接库,dll
深度学习模型与湿实验的结合,有望用于代谢通量分析
[daiy4] copy of JZ35 complex linked list
Arrangement of some library files
Agile project management of project management
Affected tree (tree DP)
Basic number theory -- Euler function
Task failed task_ 1641530057069_ 0002_ m_ 000000
Hello everyone, welcome to my CSDN blog!
Beautiful soup parsing and extracting data
Apaas platform of TOP10 abroad
How to manage the performance of R & D team?
TF coordinate transformation of common components of ros-9 ROS
Halcon clolor_ pieces. Hedv: classifier_ Color recognition
猜谜语啦(5)