当前位置:网站首页>bedtools使用教程
bedtools使用教程
2022-07-02 09:38:00 【qq_27390023】
bedtools: a powerful toolset for genome arithmetic
bedtools工具是用于广泛的基因组学分析任务的一把利器。最广泛使用的工具能够实现基因组算术:即基因组上的集合理论。例如,bedtools允许人们从广泛使用的基因组文件格式(如BAM、BED、GFF/GTF、VCF)的多个文件中交叉、合并、计数、互补和洗牌基因组区间。虽然每个单独的工具被设计用来做一个相对简单的任务(例如,与两个区间文件相交),但通过在UNIX命令行上结合多个bedtools操作可以进行相当复杂的分析。
基因组注释文件下载地址
https://genome.ucsc.edu/cgi-bin/hgTables 下载bed文件
bedtools --version # 版本号
bedtools --contact # 帮助信息
# 下载测试文件
curl -O https://s3.amazonaws.com/bedtools-tutorials/web/cpg.bed
curl -O https://s3.amazonaws.com/bedtools-tutorials/web/exons.bed
curl -O https://s3.amazonaws.com/bedtools-tutorials/web/gwas.bed
curl -O https://s3.amazonaws.com/bedtools-tutorials/web/genome.txt
###1 bedtools intersect
## 计算overlap intervals
#Tool: bedtools intersect (aka intersectBed)
#Version: v2.30.0
#Summary: Report overlaps between two feature files.
#Usage: bedtools intersect [OPTIONS] -a <bed/gff/vcf/bam> -b <bed/gff/vcf/bam>
#注:-b 可以接多个文件
# 显示cpg.bed中和exons.bed有重叠的intervals
bedtools intersect -a cpg.bed -b exons.bed
# 显示exons.bed中和cpg.bed有重叠的intervals
bedtools intersect -a exons.bed -b cpg.bed
# 同时显示重叠区域的A、B文件中的原始记录
bedtools intersect -a exons.bed -b cpg.bed -wa -wb
# 显示重叠区域的碱基数
bedtools intersect -a cpg.bed -b exons.bed -wo
# 显示每一个cpg.bed文件中的记录在exons.bed文件中的重叠记录数
bedtools intersect -a cpg.bed -b exons.bed -c
# cpg.bed文件中不和exons.bed任何intervals重叠的记录
bedtools intersect -a cpg.bed -b exons.bed -v
bedtools intersect -a cpg.bed -b exons.bed -wo
# 设定阈值,显示cpg.bed中intervals至少有50%序列和exons.bed中的重叠
bedtools intersect -a cpg.bed -b exons.bed -wo -f 0.50
# 多个文件的重叠区域
bedtools intersect -a cpg.bed -b gwas.bed exons.bed
bedtools intersect -a cpg.bed -b gwas.bed exons.bed -wa -wb -names gwas exon # 加上文件label
# sorted数据通过加-sorted参数,运行速度更快
time bedtools intersect -a exons.bed -b cpg.bed gwas.bed -sorted >>/dev/null
###2 bedtools merge
#Tool: bedtools merge (aka mergeBed)
#Version: v2.30.0
#Summary: Merges overlapping BED/GFF/VCF entries into a single interval.
#Usage: bedtools merge [OPTIONS] -i <bed/gff/vcf>
#注意:bedtools merge要求输入文件先排序
# 排序,输入文件先按染色体排序,然后按起始位置排序。
sort -k1,1 -k2,2n test.bed >test.sorted.bed
# 显示最终的"合并 "区间
bedtools merge -i exons.bed | head -n 20
# 在计算导致每个新的 "合并 "区间的重叠区间的数量时,我们将 "计算 "第一列。
bedtools merge -i exons.bed -c 1 -o count | head -n 20
# 显示所有合并成新的"合并 "区间的重叠区间的第二行
bedtools merge -i exons.bed -c 2 -o collapse | head -n 20
# 合并距离不超过1000的区间,
bedtools merge -i exons.bed -d 1000 -c 1 -o count | head -20
# 合并距离不超过90区域,分别对第一列和第四列做不同的操作
bedtools merge -i exons.bed -d 90 -c 1,4 -o count,collapse | head -20
###3 bedtools complement
#Tool: bedtools complement (aka complementBed)
#Version: v2.30.0
#Summary: Returns the base pair complement of a feature file.
#Usage: bedtools complement [OPTIONS] -i <bed/gff/vcf> -g <genome>
#注:The genome file should tab delimited and structured as follows:
# <chromName><TAB><chromSize>
# genome.txt中,exons.bed没有的区间
bedtools complement -i exons.bed -g genome.txt
###4 bedtools genomecov
#Tool: bedtools genomecov (aka genomeCoverageBed)
#Version: v2.30.0
#Summary: Compute the coverage of a feature file among a genome.
#Usage: bedtools genomecov [OPTIONS] -i <bed/gff/vcf> -g <genome>
#注:需要排序好的文件
bedtools genomecov -i exons.bed -g genome.txt
# 输出BEDGRAPH,计算intervals的depth
bedtools genomecov -i exons.bed -g genome.txt -bg | head -20
###5 bedtools jaccard
#Tool: bedtools jaccard (aka jaccard)
#Version: v2.30.0
#Summary: Calculate Jaccard statistic b/w two feature files.
# Jaccard is the length of the intersection over the union.
# Values range from 0 (no intersection) to 1 (self intersection).
#Usage: bedtools jaccard [OPTIONS] -a <bed/gff/vcf> -b <bed/gff/vcf>
# 计算相似度
bedtools jaccard -a cpg.bed -b exons.bed
###6 bedtools coverage
#Tool: bedtools coverage (aka coverageBed)
#Version: v2.30.0
#Summary: Returns the depth and breadth of coverage of features from B
# on the intervals in A.
#Usage: bedtools coverage [OPTIONS] -a <bed/gff/vcf> -b <bed/gff/vcf>
bedtools coverage -a cpg.bed -b exons.bed
参考:
http://quinlanlab.org/tutorials/bedtools/bedtools.html
边栏推荐
- The difference between SQL left join main table restrictions written after on and where
- ros gazebo相关包的安装
- Summary of data export methods in powerbi
- Jenkins installation
- Huawei game failed to initialize init with error code 907135000
- Use Huawei performance management service to configure the sampling rate on demand
- [play with FPGA learning 2 in simple terms ----- design skills (basic grammar)]
- Jinshanyun - 2023 Summer Internship
- Liftover for genome coordinate conversion
- VS2019代码中包含中文内容导致的编译错误和打印输出乱码问题
猜你喜欢
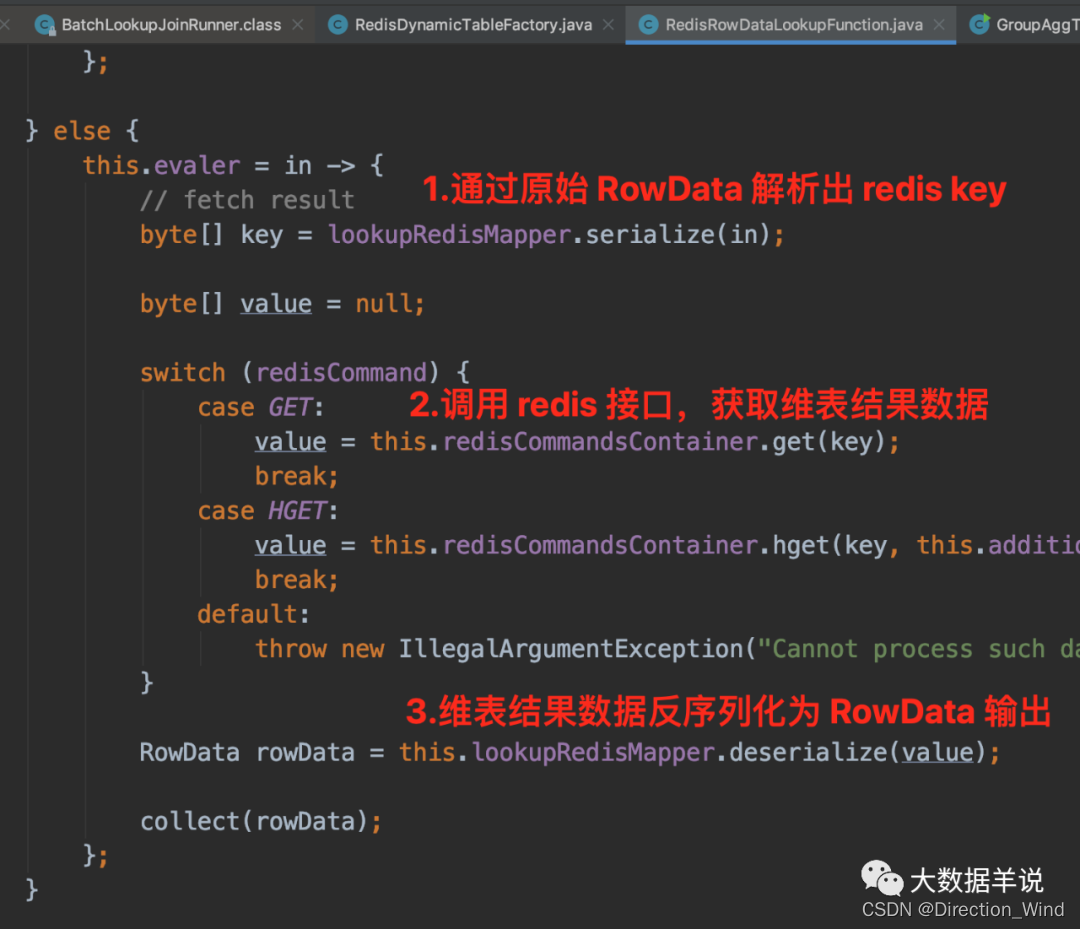
Flick two open, realized a batch lookup join (with source code)
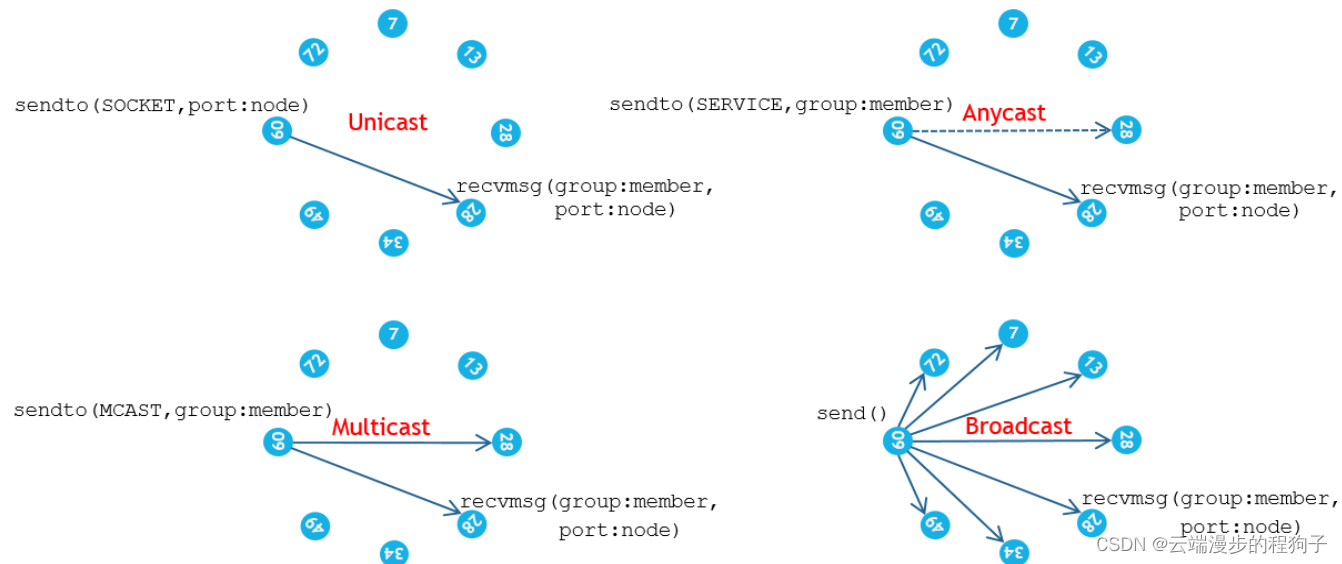
TIPC messaging3
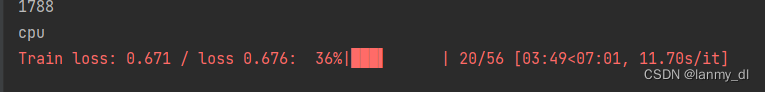
Multi line display and single line display of tqdm

2022 love analysis · panoramic report of digital manufacturers of state-owned enterprises
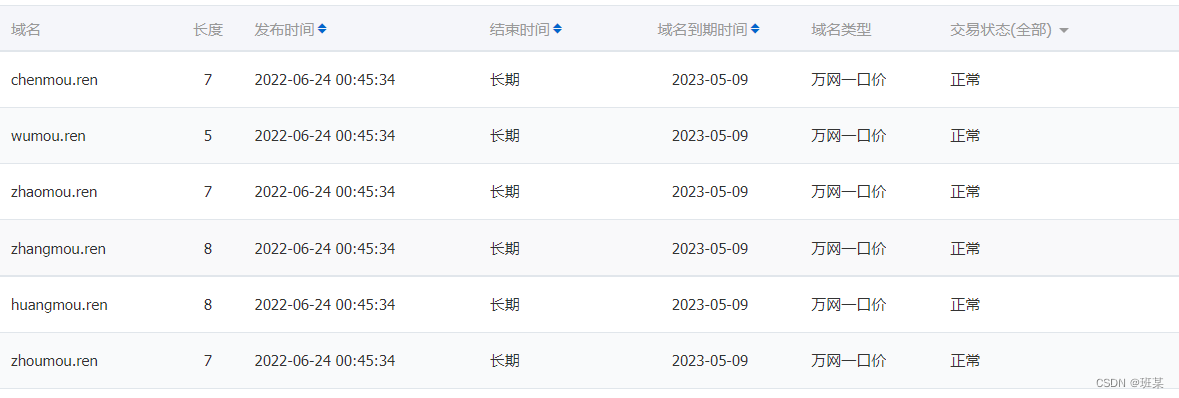
Is the Ren domain name valuable? Is it worth investing? What is the application scope of Ren domain name?

TIPC Cluster5

CentOS8之mysql基本用法

在连接mysql数据库的时候一直报错
QT learning diary 8 - resource file addition
![[AGC] how to solve the problem that the local display of event analysis data is inconsistent with that in AGC panel?](/img/66/674a06d8e45a31ae879b81554ef373.png)
[AGC] how to solve the problem that the local display of event analysis data is inconsistent with that in AGC panel?
随机推荐
MTK full dump grab
ASTParser 解析含有emum 枚举方法的类文件的踩坑记
Openmldb meetup No.4 meeting minutes
Iii. Système de démarrage et d'horloge à puce
亚马逊云科技 Community Builder 申请窗口开启
Amazon cloud technology community builder application window opens
webauthn——官方开发文档
V2x SIM dataset (Shanghai Jiaotong University & New York University)
TIPC messaging3
PHP tea sales and shopping online store
sqlite 修改列类型
函数式接口和方法引用
Indexer in C #
对毕业季即将踏入职场的年轻人的一点建议
2022 love analysis · panoramic report of digital manufacturers of state-owned enterprises
Complement (Mathematical Simulation
mmrotate旋转目标检测框架使用记录
Jenkins安装
[cloud native] 2.5 kubernetes core practice (Part 2)
tqdm的多行显示与单行显示