当前位置:网站首页>(三)R语言的生物信息学入门——Function, data.frame, 简单DNA读取与分析
(三)R语言的生物信息学入门——Function, data.frame, 简单DNA读取与分析
2022-07-06 09:17:00 【EricFrenzy】
注:本博客旨在分享个人学习心得,有不规范之处请多多包涵!
Function 函数
像其它的编程语言一样,R语言也有内置函数(如前面用到的c())和自定义函数。函数一般由三个重要的部分组成:输入参数,函数主体,返回参数。R语言的函数也是允许无输入参数或返回参数的的。以下的例子为在R语言中构造与调用函数:
#用R语言内置的function()函数来声明函数,并在括号内声明输入参数。可以用=为参数设置默认值
#getDouble为构造的函数的名称
getDouble <- function(num=1){
result <- num * 2 #函数主体
return(result) #返回参数用return()
}
myResult <- getDouble(num=20) #调用函数,myResult应为40
return()和print()的区别就在与print()会在Console显示输出而return()不会。
data.frame 数据框
比起list,data.frame在计算生物学中R语言应用更为广泛。就像matrix一样,data.frame也是二维的数据结构,即由行列组成。然而,matrix的所有元素就像vector一样一定要是同一种数据类型,但data.frame只需保证每一列的数据类型相同。这就使得data.frame能够很方便地被用来读取类似于.csv或.xlsx这样的可能有多种数据类型的表格文件。data.frame的构造请见下例:
id <- c("ENO2", "TDH3", "RPL39", "GAL4") #蛋白质的代号
protName <- c("enolase", "glyceraldehyde-3-phosphate dehydrogenase", "60S ribosomal protein L39", "regulatory protein Gal4") #蛋白质的全名
abundance <- c(24563, 22369, 16232, 32.3) #细胞中的含量,单位ppm
length <- c(437, 332, 51, 881) #蛋白质中氨基酸的数量
yeastProt <- data.frame(id, protName, abundance, length, stringsAsFactors = FALSE)
#构造data.frame;每个输入vector的长度要相同
#vector的变量名会成为表头,vector的值会按列填入data.frame
构造完成的data.frame应如下图: data.frame的索引访问与matrix类似,具体见下例:
data.frame的索引访问与matrix类似,具体见下例:
yeastProt[,1] #输出第1列的全部内容
yeastProt[2, ] #输出第2行的全部内容,包括表头
yeastProt[1, 2] #输出第1行第2列的元素,enolase
yeastProt[1:3, c(1, 3)] #输出第1-3行第1和3个元素,包括表头
yeastProt$length[1:2] #输出列标题为length的第1和第2个元素,437 332
yeastProt[1, "abundance"] #输出第1行列标题为abundance的元素,24563
yeastProt[yeastProt[, 4] > 500, 1] #输出yeastProt第4列大于500的行的第1列元素,GAL4
以下表格是vector, matrix, data.frame的互相转换:
| vector转为 | matrix转为 | data.frame转为 | |
|---|---|---|---|
| vector | \ | as.vector() | |
| matrix | as.matrix() | \ | as.matrix() |
| data.frame | as.data.frame() | as.data.frame() | \ |
在将其它数据类型转化成data.frame时,表头可能是不正确的。修改表头请见下例:
myMatrix <- matrix(data=1:12, ncol=3, nrow=4) #3x4的matrix
myDF <- as.data.frame(myMatrix) #转为data.frame
#转化完默认的列标题为V1, V2, V3... 默认行标题为1,2,3...
colnames(myDF) <- c("c1", "c2", "c3") #自定义列标题
rownames(myDF) <- c("r1", "r2", "r3", "r4") #自定义行标题
设置完成后,matrix被转变成了如下图的data.frame: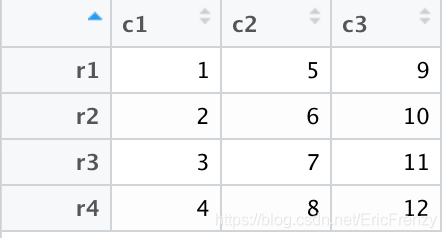
DNA序列读取与分析实例
.fasta文件是常用的读取核酸或蛋白质序列产生的文件格式。文件中开头有一些说明信息,如来源物种和染色体编号;接下来就是以单个字母代表的核酸/蛋白质编码产生的序列。本次请从这个链接下载使用NC_045512.2版本COVID19的DNA测序数据,提取码: vfti。
请看下例用R语言进行对这一序列的分析:
install.packages('seqinr', repos='http://cran.us.r-project.org') #安装第三方库
library("seqinr") #载入第三方库
covid <- read.fasta(file="covid19.fasta") #读取.fasta文件,注意路径最好写完整。读取完是list格式
covid_seq <- covid[[1]] #把完整序列单独存储为一个vector
DNA由4种base组成:A、T、C、G。A和T配对,C和G配对。根据查格夫法则(Chargaff’s law)可推导,DNA中A和T的数量相同,C和G的数量相同。因为C和G配对后由三条氢键链接,CG组合是不容易被拆散的,也就是DNA的高CG含量会减小它变异的可能性。
计算CG含量请见下例:
countTable <- table(covid_seq) #统计每个base出现的次数
cgCount <- countTable[["c"]]+countTable[["g"]] #C和G的数量
totalCount <- length(covid_seq) #序列的总长
cgContent <- cgCount/totalCount #CG在序列中的占比
另一个比较重要的概念是DNA word,即长度大于1个base的DNA组合。通过分析DNA word是否被过表达或低表达,可以在一定程度上推测该物种演化的轨迹。 ρ ρ ρ用来表示一个DNA word是否被过表达或低表达。对于一个长度为2的DNA word,计算公式如下:
ρ ( x y ) = f x y f x ⋅ f y ρ(xy) = \frac{f_{xy}}{f_{x}·f_{y}} ρ(xy)=fx⋅fyfxy
其中 f x y f_{xy} fxy是DNA word的出现频率除以序列总长, f x f_{x} fx和 f y f_{y} fy是单个base的出现频率除以序列总长。计算某DNA word被如何表达请见下例:
baseCount <- count(covid_seq, 1) #用seqinr这个第三方库中的count()函数找长度为1的base的出现次数,和R内置的table()类似
wordCount <- count(covid_seq, 2) #找长度为2的DNA word的出现次数
totalLength <- length(covid_seq) #序列总长
#比如,我们要找TT这个DNA word的出现次数
rouTT <- (wordCount[["tt"]]/totalLength)/((baseCount[["t"]]/totalLength)*(baseCount[["t"]]/totalLength))
当 ρ ρ ρ等于1时,这个DNA word为完全随机出现,与单个base出现概率的乘积相同。然而,当 ρ ρ ρ大于1时,它的出现概率大于随机出现的概率,说明它被过表达,所以可推测这个DNA word在这一物种的进化方面可能有某些有益作用。反之亦然。
结束语
下次会以分子生物学和生态学的数据为例,介绍R语言中的矩阵图、条形统计图、饼图、点阵图与线性回归、带状图的使用方法,敬请期待!有任何问题或想法欢迎留言和评论!
边栏推荐
- Missing value filling in data analysis (focus on multiple interpolation method, miseforest)
- Characteristics, task status and startup of UCOS III
- JS object and event learning notes
- E-commerce data analysis -- User Behavior Analysis
- Unit test - unittest framework
- 高通&MTK&麒麟 手機平臺USB3.0方案對比
- Raspberry pie tap switch button to use
- GNN的第一个简单案例:Cora分类
- VSCode基础配置
- Pytorch实现简单线性回归Demo
猜你喜欢
Common properties of location
Basic operations of databases and tables ----- modifying data tables
ES6 grammar summary -- Part I (basic)
E-commerce data analysis -- User Behavior Analysis

RT-Thread 线程的时间片轮询调度
open-mmlab labelImg mmdetection
A possible cause and solution of "stuck" main thread of RT thread

RT thread API reference manual
【ESP32学习-2】esp32地址映射
Kaggle competition two Sigma connect: rental listing inquiries (xgboost)
随机推荐
Pytoch temperature prediction
Types de variables JS et transformations de type communes
I2C bus timing explanation
Togglebutton realizes the effect of switching lights
VSCode基础配置
arduino获取随机数
Mp3mini playback module Arduino < dfrobotdfplayermini H> function explanation
Arduino uno R3 register writing method (1) -- pin level state change
Knowledge summary of request
Common properties of location
1081 rational sum (20 points) points add up to total points
几个关于指针的声明【C语言】
vim命令行笔记
js题目:输入数组,最大的与第一个元素交换,最小的与最后一个元素交换,输出数组。
STM32 how to locate the code segment that causes hard fault
Programmers can make mistakes. Basic pointers and arrays of C language
Basic operations of databases and tables ----- view data tables
gcc 编译选项
Esp8266 uses Arduino to connect Alibaba cloud Internet of things
Symbolic representation of functions in deep learning papers