当前位置:网站首页>(五)R语言入门生物信息学——ORF和序列分析
(五)R语言入门生物信息学——ORF和序列分析
2022-07-06 09:17:00 【EricFrenzy】
注:本博客旨在分享个人学习心得,有不规范之处请多多包涵!
概念介绍
在人体内,为了表达DNA上的基因,这个基因包含的DNA在被转录为pre-mRNA后经过进一步处理成为成熟的mRNA,mRNA紧接着会被核糖体用来合成蛋白质,从而控制生物体的反应。在mRNA上,每三个碱基组成一个密码子,对应一种氨基酸。下图为密码子与氨基酸的对照表: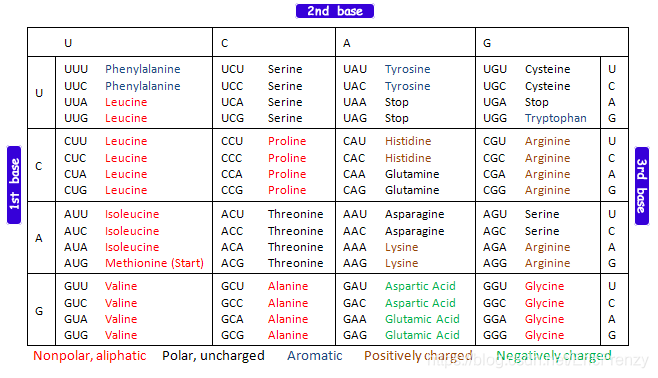
要合成一个正常的蛋白质,mRNA序列上的两端需要分别有一个起始密码子(图上标为start)和一个终止密码子(图上标为stop)。但在DNA上有许多这样子的起始和终止密码子,产生很多种不同的序列组合。为了在DNA上找到所有可能的能用来制作某种蛋白质的序列组合,我们用开放阅读框(ORF,Open Reading Frame)来找到所有具有编码蛋白质潜能的序列。
找ORF的代码实现
在R语言中找ORF的程序流程如下: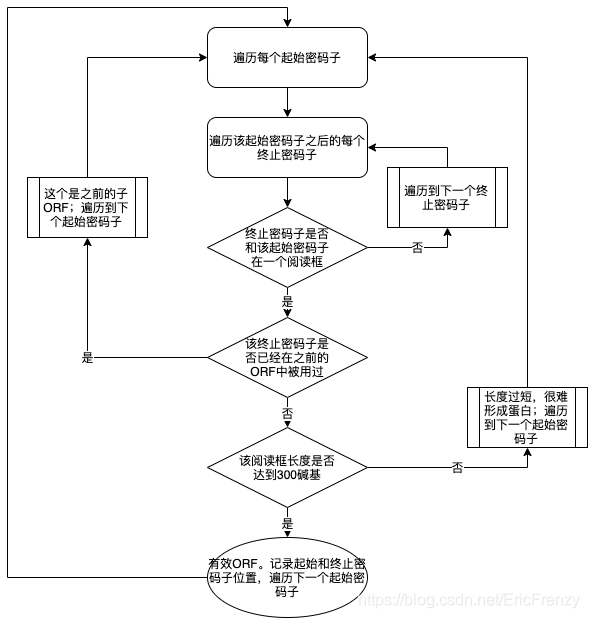
下面是具体代码:
findORF <- function(seq){
#传入参数为DNA序列,注意方向一定要是5'到3'
findStartCodons <- function(seq){
#找起始密码子的函数
startcodons <- numeric(0) #创建空函数
k <- 1
for(i in 1:(length(seq)-5)){
#以密码子的第一位碱基位置计算,最后五位无需检查,因为长度过短
if(seq[i] == "a" && seq[i+1] == "t" && seq[i+2] == "g"){
#ATG对应起始密码子
startcodons[k] <- i #记录位置
k <- k + 1 #位置下标加一
}
}
return(startcodons) #返回结果
}
findStopCodons <- function(seq){
#找终止密码子的函数
stopcodons <- numeric(0) #创建空函数
k <- 1
for(i in 1:(length(seq)-2)){
#以密码子的第一位碱基位置计算
if((seq[i] == "t" && seq[i+1] == "a" && seq[i+2] == "a") || (seq[i] == "t" && seq[i+1] == "a" && seq[i+2] == "g") || (seq[i] == "t" && seq[i+1] == "g" && seq[i+2] == "a")){
#TAA TAG TGA对应终止密码子
stopcodons[k] <- i #记录位置
k <- k + 1 #位置下标加一
}
}
return(stopcodons) #返回结果
}
startcodon <- findStartCodons(seq) #找到所有的起始密码子
stopcodon <- findStopCodons(seq) #找到所有的终止密码子
usedStop <- numeric(0) #记录用过的终止密码子
ORFs <- character(0) #记录有效开放阅读框
k <- 1
for(i in startcodon){
#遍历所有起始密码子
for(j in stopcodon){
#遍历所有终止密码子
if((j-i)%%3==0 && j > i){
#如果在一个阅读框内,即两个密码子之间的位置为3的整数
if(j %in% usedStop){
#如果终止密码子被用过
break #跳出这次循环,到下一个起始密码子
}else if(j-i < 300){
#如果密码子之间的序列长度过短
break #同上
}else{
ORFs[k] <- paste(i, "to", j) #生成字符串,记录的结果如"1 to 3001"
usedStop[k] <- j #记录用过的终止密码子
k <- k + 1 #位置下标加一
break #跳出本次循环,到下一个起始密码子
}
}
}
}
return(ORFs) #返回结果
}
这种找ORF的算法比较简单快速,但相应地准确度会有所下降。在NCBI官网有更准确的算法。
结束语
在找到ORF后,就能将该ORF与数据库中的已知序列进行比对,从而预测该物种基因的组成与功能等有用信息。下次将会介绍Needleman-Wunsch这一序列全局比对算法,敬请期待!和有任何问题或想法欢迎留言和评论!
边栏推荐
猜你喜欢

Symbolic representation of functions in deep learning papers
Arduino uno R3 register writing method (1) -- pin level state change
Amba, ahb, APB, Axi Understanding

Kconfig Kbuild
Comparison of solutions of Qualcomm & MTK & Kirin mobile platform USB3.0

level16
![Several declarations about pointers [C language]](/img/9b/ace0abbd1956123a945a98680b1e86.png)
Several declarations about pointers [C language]
arduino JSON数据信息解析
PyTorch四种常用优化器测试

Reno7 60W super flash charging architecture
随机推荐
Pytorch-温度预测
冒泡排序【C语言】
嵌入式启动流程
ES6 grammar summary -- Part 2 (advanced part es6~es11)
RuntimeError: cuDNN error: CUDNN_ STATUS_ NOT_ INITIALIZED
Priority inversion and deadlock
优先级反转与死锁
Basic knowledge of lithium battery
C language callback function [C language]
列表的使用
Kaggle competition two Sigma connect: rental listing inquiries (xgboost)
Apprentissage automatique - - régression linéaire (sklearn)
数据分析之缺失值填充(重点讲解多重插值法Miceforest)
Basic operations of databases and tables ----- view data tables
JS regular expression basic knowledge learning
JS object and event learning notes
ESP8266通过arduino IED连接巴法云(TCP创客云)
Redis cache update strategy, cache penetration, avalanche, breakdown problems
Page performance optimization of video scene
[golang] leetcode intermediate - fill in the next right node pointer of each node & the k-smallest element in the binary search tree