当前位置:网站首页>2020 Bioinformatics | GraphDTA: predicting drug target binding affinity with graph neural networks
2020 Bioinformatics | GraphDTA: predicting drug target binding affinity with graph neural networks
2022-07-06 13:50:00 【发呆的比目鱼】
2020 Bioinformatics | GraphDTA: predicting drug target binding affinity with graph neural networks

Paper: https://academic.oup.com/bioinformatics/article/37/8/1140/5942970?login=false
Code:https://github.com/thinng/GraphDTA
摘要
新药的开发成本高、耗时长,而且往往伴随着安全问题。药物再利用可以通过为已批准的药物寻找新用途来避免昂贵且冗长的药物开发过程。为了有效地重新利用药物,了解哪些蛋白质被哪些药物靶向是有用的。估计新药物-靶点对相互作用强度的计算模型有可能加快药物再利用。已经为这项任务提出了几个模型。然而,这些模型将药物表示为字符串,这不是表示分子的自然方式。我们提出了一个名为GraphDTA的新模型它将药物表示为图形,并使用图形神经网络来预测药物与靶点的亲和力。我们表明,图神经网络不仅比非深度学习模型更好地预测药物-目标亲和力,而且优于竞争的深度学习方法。我们的结果证实,深度学习模型适用于药物-靶点结合亲和力预测,并且将药物表示为图形可以导致进一步的改进。
介绍
药物-靶标亲和力 (DTA) 预测计算方法有几种方法:
- 分子对接,它通过评分函数预测药物-靶标复合物的稳定 3D 结构。
- 使用协同过滤。例如,SimBoost模型使用药物之间和目标之间的亲和力相似性来构建新特征。
- 使用在药物和蛋白质序列的一维表示上训练的神经网络。例如,DeepDTA模型使用一维表示和一维卷积层(带有池化)来捕获数据中的预测模式
药物表征
SMILES可通过rdkit开源软件生成graph的形式,然后通过图卷积网络表示学习得到药物特征向量。其中每个节点是一个多维01特征向量,表达了五条信息:原子符号、相邻原子个数、相邻氢原子个数、原子的隐含值、原子是否处于芳香结构中。
蛋白表征
由于蛋白表示图结构比较困难, 蛋白质结果特征以one-hot编码表示。目标的基因名称从 UniProt 数据库中获取蛋白质序列。该序列是一串代表氨基酸的 ASCII 字符。每种氨基酸类型根据其相关的字母符号用一个整数编码[例如,丙氨酸 (A) 为 1,胱氨酸 为 3,天冬氨酸 (D) 为 4,依此类推],使蛋白质可以表示为一个整数序列。
分子图模型结构
作者提出了一种新的基于图神经网络和传统 CNN 的 DTA 预测模型。如下图所示。首先对蛋白质序列进行分类编码,然后将嵌入层添加到序列中,其中每个(编码)字符由 128 维向量表示。接下来,使用三个 1D 卷积层从输入中学习不同级别的抽象特征。最后,应用最大池化层来获得输入蛋白质序列的表示向量。这种方法类似于现有的基线模型。对于药物,我们使用分子图并试验了四种图神经网络变体,包括 GCN ( Kipf and Welling, 2017 )、GAT ( Veličković et al., 2018 ))、GIN ( Xu et al., 2019 ) 和组合的 GAT-GCN 架构。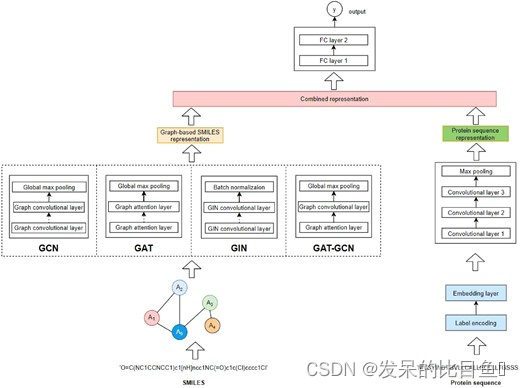
实验与结果
研究人员主要通过对比非深度学习模型与比较流行的深度学习模型,通过测量计算一致性指数CI(指示预测值与实际值的一致性)与均方误差MSE这两个指标来表示模型的好坏。为了使实验结果具有比较性,分别在Davis与Kiba数据集对模型进行测量。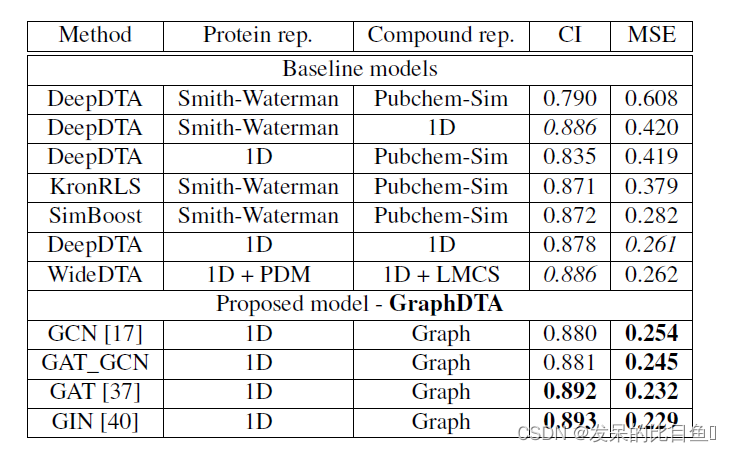
Davis数据集模型测量结果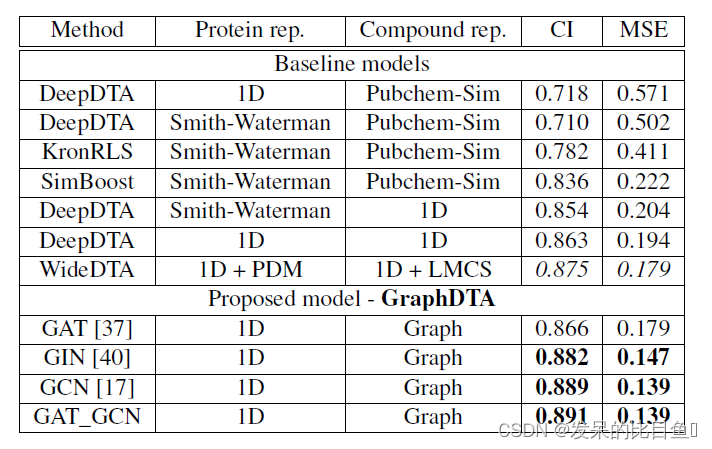
两种数据集中的测量结果都表示在基于GAT-GCN结合的图表示模型中预测性能最佳。
结论
本项工作中,研究人员提出了一种计算药物-靶标结合亲和力的新方法,称为GraphDTA;旨在降低药物开发的难度,减少发现新药物靶标相互作用在时间与成本上的花费,缩短药物开发周期。该模型使用由SMILES数据重构得来的二维图结构数据,能够表达药物的较完整信息,因此该方法能够获得较好的预测性能。
参考
边栏推荐
- JPEG2000-Matlab源码实现
- [Digital IC manual tearing code] Verilog automatic beverage machine | topic | principle | design | simulation
- Explain ESM module and commonjs module in simple terms
- Five wars of Chinese Baijiu
- Quick access to video links at station B
- 强化学习-学习笔记5 | AlphaGo
- GPS从入门到放弃(十八)、多路径效应
- GPS from getting started to giving up (16), satellite clock error and satellite ephemeris error
- Make menuconfig has a recipe for target 'menuconfig' failed error
- Shake Sound poussera l'application indépendante de plantation d'herbe "louable", les octets ne peuvent pas oublier le petit livre rouge?
猜你喜欢

红杉中国,刚刚募资90亿美元
基于LM317的可调直流电源

Five wars of Chinese Baijiu

【MySQL】Online DDL详解
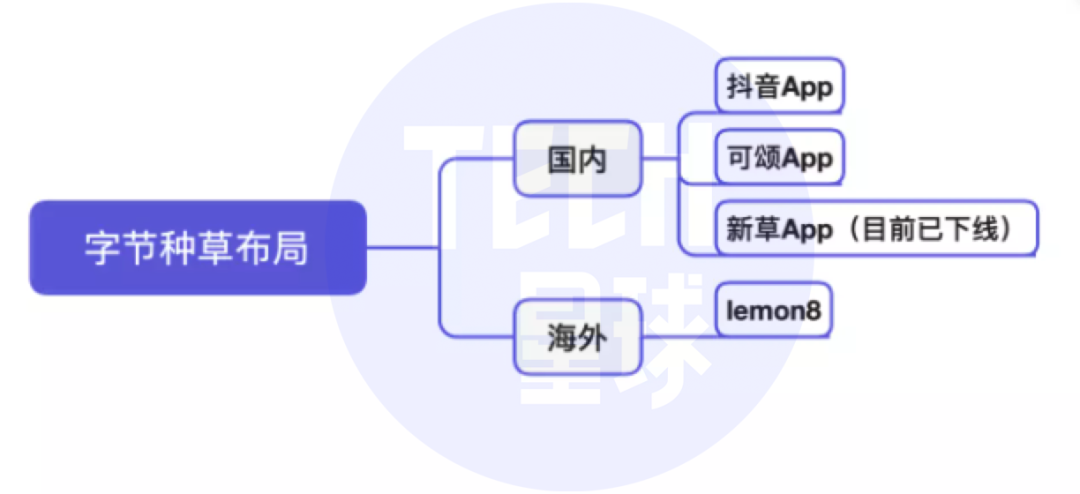
Tiktok will push the independent grass planting app "praiseworthy". Can't bytes forget the little red book?

GPS from getting started to giving up (16), satellite clock error and satellite ephemeris error
PostgreSQL 修改数据库用户的密码

It's not my boast. You haven't used this fairy idea plug-in!
Bat script learning (I)
![[Chongqing Guangdong education] Tianjin urban construction university concrete structure design principle a reference](/img/61/976c7d86ab3b2df5f5af3beefbf547.png)
[Chongqing Guangdong education] Tianjin urban construction university concrete structure design principle a reference
随机推荐
C语言:#if、#def和#ifndef综合应用
在Pi和Jetson nano上运行深度网络,程序被Killed
The relationship between root and coefficient of quadratic equation with one variable
Redistemplate common collection instructions opsforhash (IV)
The golden age of the U.S. technology industry has ended, and there have been constant lamentations about chip sales and 30000 layoffs
Tips for web development: skillfully use ThreadLocal to avoid layer by layer value transmission
Run the deep network on PI and Jetson nano, and the program is killed
LeetCode学习记录(从新手村出发之杀不出新手村)----1
From campus to Tencent work for a year of those stumbles!
【sciter】: 基于 sciter 封装通知栏组件
[Chongqing Guangdong education] Information Literacy of Sichuan Normal University: a new engine for efficiency improvement and lifelong learning reference materials
Write a rotation verification code annotation gadget with aardio
make menuconfig出现recipe for target ‘menuconfig‘ failed错误
Broadcast variables and accumulators in spark
Numpy download and installation
JPEG2000 matlab source code implementation
GPS从入门到放弃(十三)、接收机自主完好性监测(RAIM)
Shell product written examination related
Codeforces Round #274 (Div. 2) –A Expression
[Li Kou brushing questions] one dimensional dynamic planning record (53 change exchanges, 300 longest increasing subsequence, 53 largest subarray and)