当前位置:网站首页>Chapter 1 :Application of Artificial intelligence in Drug Design:Opportunity and Challenges
Chapter 1 :Application of Artificial intelligence in Drug Design:Opportunity and Challenges
2022-07-06 08:49:00 【UniversalNature】
reading notes of《Artificial Intelligence in Drug Design》
文章目录
Chapter 1 Application of Artificial intelligence in Drug Design : Opportunity and Challenges
1.Introduction : What Challenges Does Drug Design Face
- R&D cost increased dramatically.
 - Hits with promising potency and ADMET properties are chosen as “lead compounds” —— these then need to be optimized for potency and selectivity while maintaining an appropriate ADMET profile. This process is often ineffective at finding molecules with the right pharmacodynamic and pharmacokinetic properties in patients.- Datasets problem:
- Hits with promising potency and ADMET properties are chosen as “lead compounds” —— these then need to be optimized for potency and selectivity while maintaining an appropriate ADMET profile. This process is often ineffective at finding molecules with the right pharmacodynamic and pharmacokinetic properties in patients.- Datasets problem:
- Many datasets are disproportionately focused on a small range of well-studied endpoints.
- Datasets describing in vitro activities of compounds is larger than those describing their in vivo effects.
- Consistent annotation of data is challenging.
- There is little hope of conducting simulations of complex physiological systems, necessitating the use of empirical models.
2. Application of Artificial Intelligence in Drug Design
2.1.Virtual Screening
2.1.1.Introduction
- HTS(high-throughput screening) screens large chemical libraries against project relevant activity assays.
- VS(virtual screening) attempts to address the shortcomings of HTS by screening compounds in silico rather than in vitro.
- VS can be categorized into two types: ligand-based VS and structure-based VS.
- ligand-based VS uses a set of compounds that are known to be active and tries to identify other molecules. This needs a predictive model to prioritize active compounds.
- structure-based VS evaluates a ligand based on the complementarity of 3D structures with the target’s binding pockets. This needs 3D information which is hard to obtain.
2.1.2.Dataset Bias in Machine Learning-Based Virtual Screening
- Molecular datasets used in VS incorporate bias due to several reasons.
- Their size is comparably small relative to the space of potential molecules.
- The nature of drug development pipelines leads to bias : synthesis efforts typically focus around known successful molecules.
- Novel molecules are often proposed in series that grow during hit selection and lead optimization. As a result, explored regions of chemical space are formed of clusters rather than uniform samples.
- The practitioners should be careful about how they split their dataset for training and testing. (Butina-Taylor)
- Another caveat that practitioners should be aware of is the lack of universally accepted chemical datasets on which to evaluate models for VS. (DUD-E, MUV, AVE)
- Bias is not always undesirable.
2.1.3.Receptor Structure-Based Virtual Screening (Docking)
- Most ML approaches to structure-based VS have focused on improving the scoring function. Gnina and AtomNet have develop their own standalone scoring functions. Other ML model (such as △VinaRF) for scoring attempt to improve rather than replace.
- One way to improve scoring functions is to tailor them to the problem of interest. ML model is more suitable than traditional docking algorithms.
- An important limitation of current ML model with respect to traditional docking algorithms is that they generally lack the capabilities to produce docking poses, so they rely on external software to obtain them. This means ML model performance is capped by the performance of external pose generation.
2.1.4.Ligand-Based Methods (QSAR)
What characterizes ligand-based VS is that it does not utilize any information about the receptor. Therefore, it can be applied to general problems.
QSAR and ML model is similar: both are supervised methods that identify pattern in molecular data to learn a target signal.
In addition to fitting complicated target functions, ML model can allows a greater breadth of molecular representations, some of them is very abstract but important such as molecule-induced transcriptomic signatures or cell painting imaging profiles.
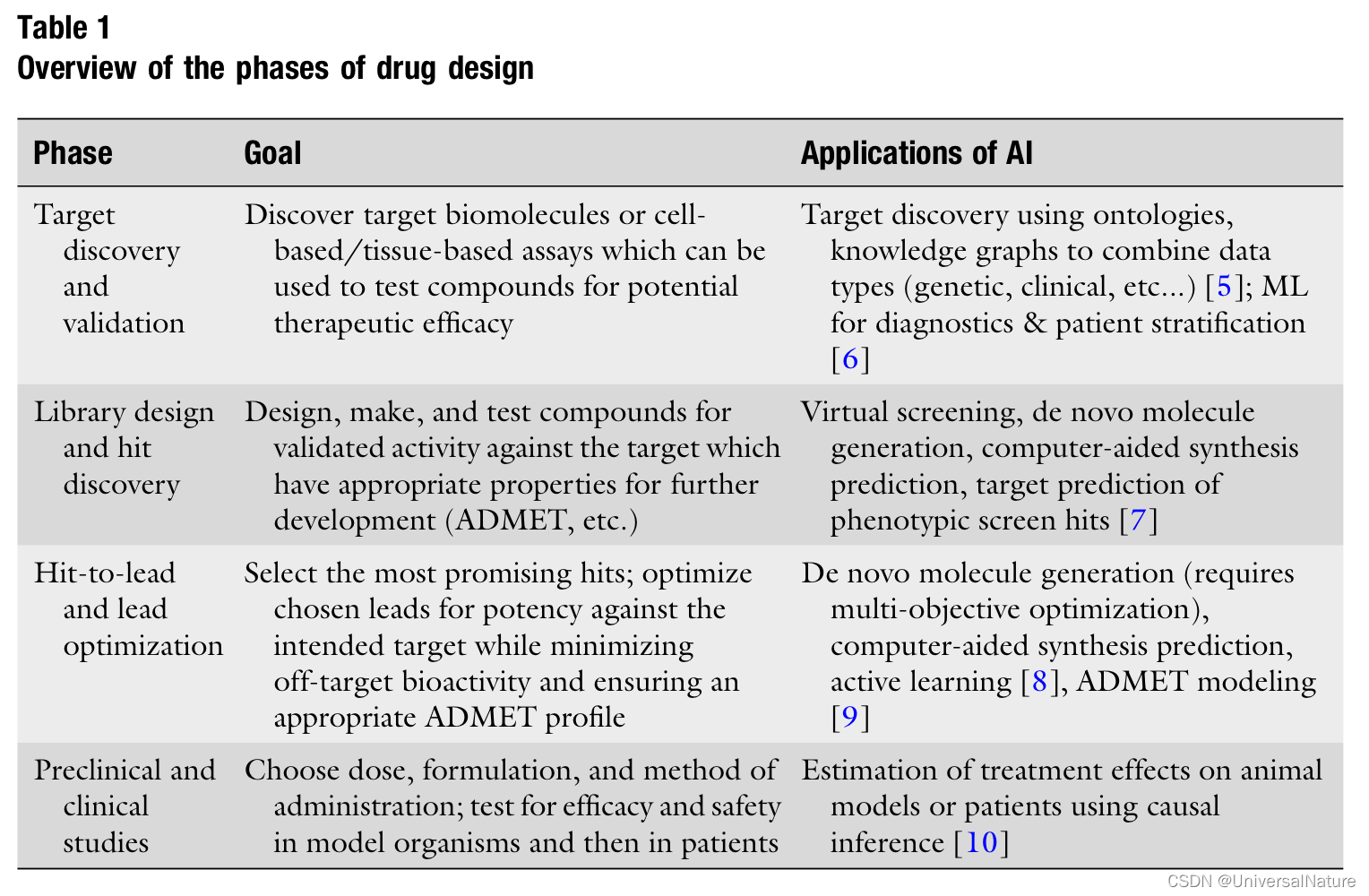
In addition to using new biological data types, another advantage of some ML model is that they can learn their own representation from a dataset. VAE’s latent space is usually smaller in size than the input, so the model is forced to find a compressed representation of the input.
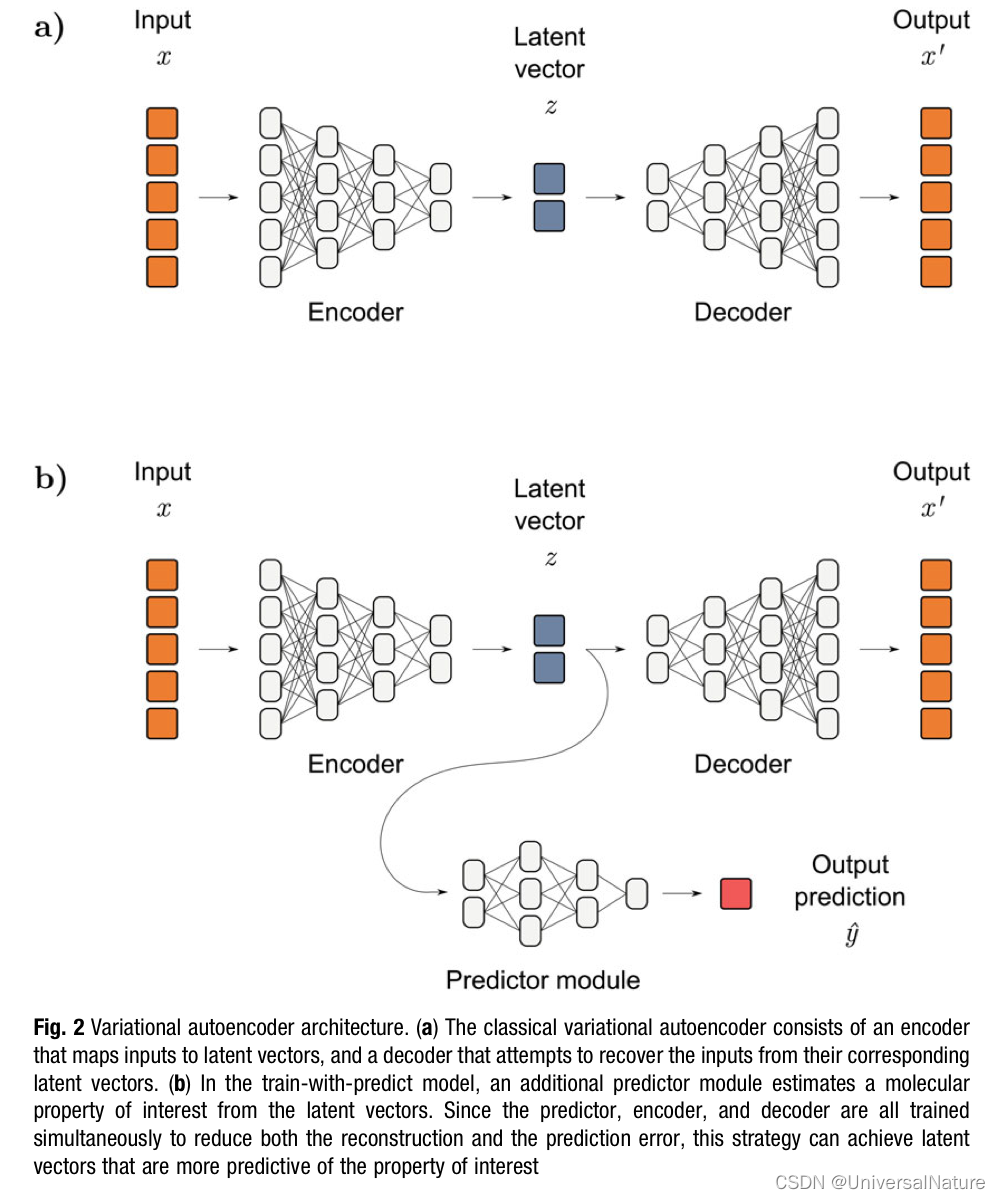
Some ML models accepted flexible training regimes which presents another opportunity for improvement of ligand-based. There are several methods based on the idea of shared knowledge, such as transfer learning, multitask learning, self-supervised learning.
Flexibility brings practical difficulties, too. The high degree of choice makes it difficult to decide what model is most promising for a specific problem. The part of challenge is that it is difficult to diagnose why a ML method is successful.
2.1.5.Hybrid VS (Incomprehensible)
- Hybrid VS refer to statistic al models for bioactivity prediction that incorporate information about the receptor, but in more abstract form than a geometric description of the binding pocket. It is often considered an expansion of ligand-based VS and QSAR.
- The most common type of Hybrid VS are proteochemometirc models (PCM).
- Many of the opportunities and limitations discussed in Ligand-Based VS is applicable here.
- PCM is the flexibility and customizability of input representations. DeppDTA and DGraphDTA could produce target representations.
- PCM is building models that scale to the big-data regime, such as collaborative filtering.
- ML is helpful for PCM in uncertainty quantification. A work that applied a Gaussian process to PCM found that Bayesian model achieved calibrated variance estimates for each prediction.
- ML could be used to design novel hybrid workflows different from PCM, such as DeepDocking.
边栏推荐
猜你喜欢

opencv+dlib实现给蒙娜丽莎“配”眼镜
After PCD is converted to ply, it cannot be opened in meshlab, prompting error details: ignored EOF

同一局域网的手机和电脑相互访问,IIS设置
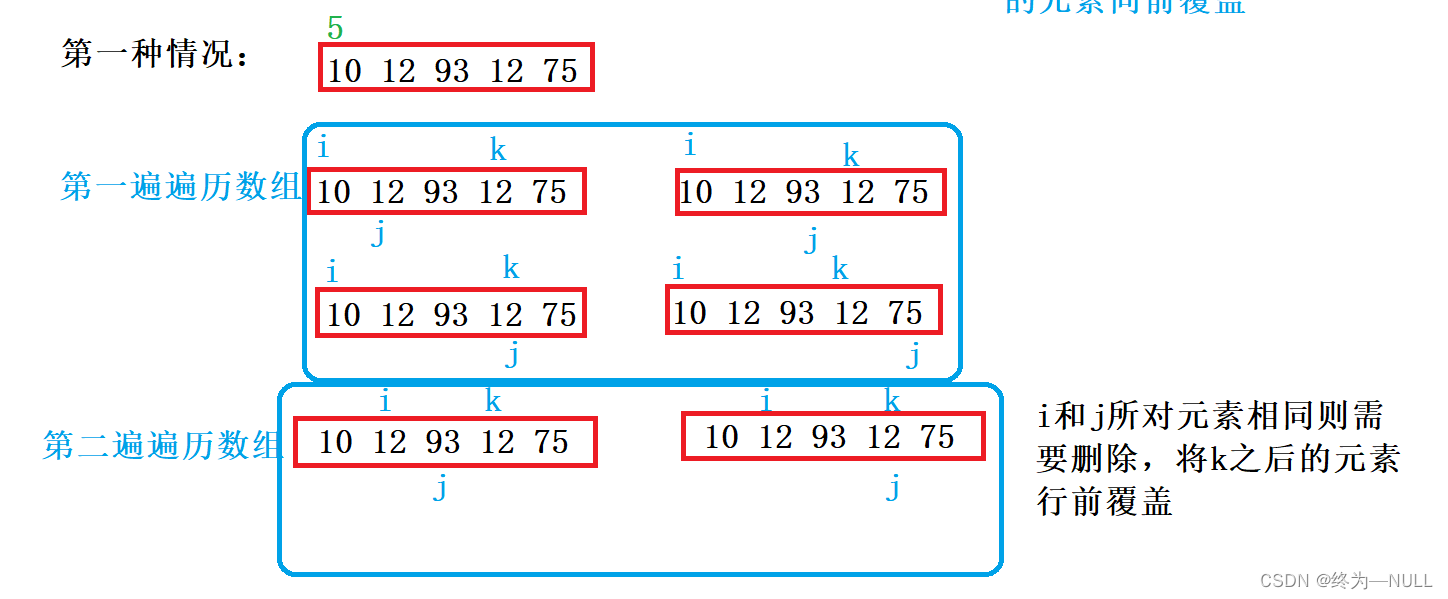
C语言双指针——经典题型
UnsupportedOperationException异常
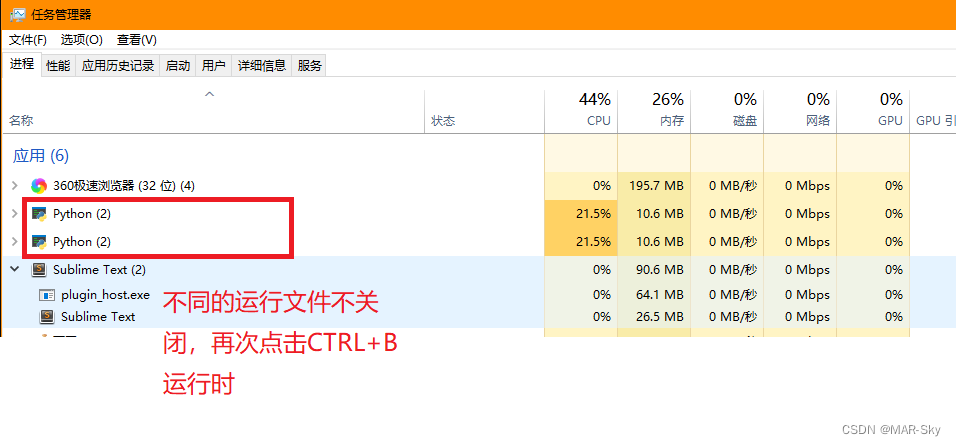
sublime text没关闭其他运行就使用CTRL+b运行另外的程序问题
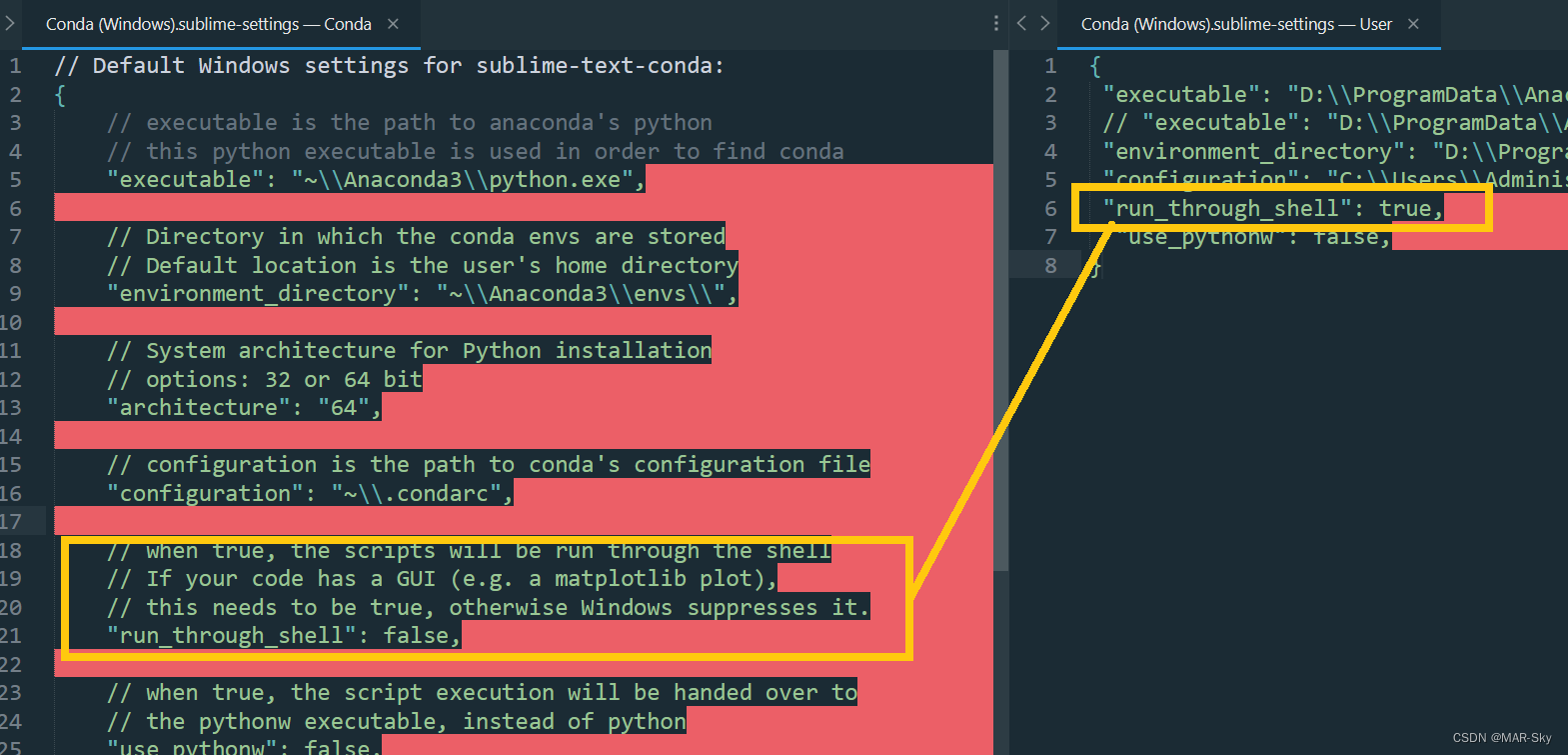
Sublime text in CONDA environment plt Show cannot pop up the problem of displaying pictures

深度剖析C语言指针
项目连接数据库遇到的问题及解决
LeetCode:236. 二叉树的最近公共祖先
随机推荐
Shift Operators
LeetCode:41. 缺失的第一个正数
Generator parameters incoming parameters
软件压力测试常见流程有哪些?专业出具软件测试报告公司分享
Visual implementation and inspection of visdom
JS inheritance method
UML图记忆技巧
JS native implementation shuttle box
opencv+dlib实现给蒙娜丽莎“配”眼镜
Function coritization
CSP first week of question brushing
View computer devices in LAN
The harm of game unpacking and the importance of resource encryption
Navicat premium create MySQL create stored procedure
@JsonBackReference和@JsonManagedReference(解决对象中存在双向引用导致的无限递归)
sublime text的编写程序时的Tab和空格缩进问题
pytorch查看张量占用内存大小
按位逻辑运算符
Variable length parameter
如何进行接口测试测?有哪些注意事项?保姆级解读